Peters综合征临床特征和基因突变的研究
2016-12-05黄丽琴卢国华毛平安孙新成
黄丽琴,卢国华,谢 阳,毛平安,孙新成,孟 永
·临床研究·
Peters综合征临床特征和基因突变的研究
黄丽琴1,卢国华1,谢 阳1,毛平安1,孙新成1,孟 永2
•METHODS: Ten congenital corneal opacities affected patients were enrolled from our pediatric and genetic eye clinic. Medical and ophthalmic histories were obtained. Genomic DNA was prepared from venous leukocytes after informed consent conforming was obtained from each participant. The coding regions and the flanking exon-intron junctions of this gene were amplified by polymerase-chain reaction (PCR) and subsequently analyzed by direct sequencing. Variations detected were further evaluated in 100 normal controls by direct sequencing.
•RESULTS: One affected individual characterized by systemic changes such as congenital heart anomalies and hearing loss showed the consistent phenotypes with Peters syndrome. Sequence analysis of the PITX2 gene revealed one novel mutation, c. 788G>A. Nucleotide sequence analysis showed that this mutation led to the functional abnormal of this gene, however, no mutation was observed in any unaffected member or 100 normal unrelated individuals.
•CONCLUSION: A novel mutation in the PITX2 gene have been identified, this is the first report on a mutation in a Chinese Peters syndrome and the result expand the mutation spectrum of PITX2, further clarify the clinical features of the disease. All these will be useful foundations for clinical diagnosis, medical therapy and pathogenesis.
目的:分析国人Peters综合征的临床特征,并确定致病基因突变,为该病的临床诊断和治疗及发病原因提供依据。
方法:选取2012/2015年在常州市第二人民医院眼科就诊的10例先天性角膜混浊的患者,并收集详细的相关临床资料。征得患者及其家系成员的同意后抽血制备基因组DNA,用聚合酶链反应(PCR)对致病基因PITX2的编码区及其临接内含子进行扩增后,直接测序分析该基因。同时检测100位无亲缘关系的正常人外周血标本进行对照验证。
结果:患者1例的临床特点包括先天性角膜中央部混浊白斑,伴有相应区域的角膜后部基质变薄和后弹力层缺损,且患者伴有全身系统如心脏和听力损害等改变,符合Peters综合征的临床诊断;该患者PITX2基因突变筛查结果发现了1种新突变,c. 788G>A,导致该基因的功能异常,而家属中表型正常者及无亲缘关系的正常对照者均未发现该基因突变。
结论:先天性角膜混浊患者10例中检测到1个新PITX2基因突变,符合Peters综合征的临床诊断,这是中国首次报道Peters综合征的PITX2基因突变,结果丰富了PITX2基因突变频谱,并进一步明确了Peters综合征的临床特点,为该病的临床诊断和治疗及发病原因提供了依据。
Peters综合征;先天性角膜混浊;PITX2基因;突变
引用:黄丽琴,卢国华,谢阳,等.Peters综合征临床特征和基因突变的研究.国际眼科杂志2016;16(12):2237-2240
0引言
Peters综合征(简称PPS)最初在20世纪由Peter依据临床表现及组织学特点进行描述,是最常见的一种先天性角膜混浊眼病。国外报道在新生儿中的发病率为1.2/100 000,其大多数是散发病例,但也有一些染色体显性及隐性遗传的报道。PPS是临床中少见病,且易与其他引起先天性角膜混浊的眼病如巩膜化角膜、角膜皮样瘤、先天性青光眼、小眼畸形、产伤性角膜白斑及黏多糖贮积症眼病等相混淆。国外已有较多关于本病分子遗传学的研究,在国内,目前尚缺少该眼病的的致病基因的研究及临床特征的描述,以致很多临床医师没有足够认识。本研究收集10例先天性角膜混浊病患者的临床资料,明确PPS的临床特点,利用PCR和直接测序方法对其致病基因PITX2进行突变检测,了解PPS基因型与表型的关系,为该病的临床诊断和治疗及发病原因提供依据。
表1 PCR反应所需引物的序列和相应扩增产物的长度与退火温度

基因外显子正向引物(5 -3 )反向引物(5 -3 )产物长度(bp)退火温度(℃)PITX21GGCCGCCGCTTCTTACACACTGGCGATTTGGTTCTGATTT254632CTGCCTCGCCCCTCCCCCTCCTCTTCAAGCAGCAGGCGCGTCAGGTC350643TCTTCGTTTGCCCGCTGACTTTGGCGAACGACCACTCCCACCAC549634GGCGCGGACACCCACACTTGGCGCTGCCTTCCACATTCTCT487655-1CTGGCCCTGGTATCTTGGTGTGCGGTGGATAGGGAGGCGGATGTAAG346635-2CGACGACATGTACCCAGGCTATTCCGACGGGCTACTCAGGTTGTTCA256645-3TCCCGGGCTCCAGTCTCAACAGTTTCTTTAGTGCCCACGACCTTCT35564
注:外显子5共设计三对引物进行PCR扩增;PCR反应体系中PITX2的GC含量较高,故引物采用2×GC Buffer I(Takara)的缓冲液。
1对象和方法
1.1对象 选取先天性角膜混浊眼病临床标准:(1)先天性角膜混浊白斑;(2)患眼角膜无血管化;(3)可伴有其它眼部异常:包括小角膜、小眼球、巩膜化角膜、中央凹发育不良、视乳头发育不良、青光眼、白内障、永存原始玻璃体增生症等;(4)可伴有全身异常:身材短小、听力损害、心脏疾病等。符合以上标准的共有10个没有亲缘关系的初步诊断为先天性角膜混浊的患者参加本项研究,详细询问所有受试者病史,并行眼部检查以及全身检查。遵循相关伦理原则,征得患者及其家系成员的同意后抽血制备基因组DNA,并收集相关临床资料。100例正常对照者均无已知眼遗传病及全身遗传病,与患者无亲缘关系。
1.2方法
1.2.1聚合酶链反应扩增 采集10例先天性角膜混浊患者肘静脉血5mL,用常规酚氯仿方法,从患者和100例正常人外周血中提取DNA。根据Genbank中人PITX2基因cDNA序列,采用Primer 5.0软件设计引物(表1)。采用聚合酶链反应(PCR)扩增PITX2基因所有外显子和临近内含子。PCR反应体系为20μL,包含模板DNA 40ng/μL,2×GC缓冲液12.5μL,引物(上海生工公司)0.4μL×2,2.5mmol/L dNTP 0.6μL,rTaq酶0.4μL。PCR反应条件:94℃预变性5min,94℃变性30s,63℃~65℃退火30s,72℃延伸30s,30~34个循环,反应终止后72℃后延伸5min,4℃保存。琼脂糖电泳:应用15g/L琼脂糖凝胶水平电泳检测PCR产物,在紫外灯下观察是否有电泳条带、产物量多少和是否有非特异性扩增。
1.2.2突变基因筛查 对PCR产物纯化,使用AB13730 DNA全自动测序仪上机进行双向测序,测序结果采用Lasegen软件进行对比分析,结果与Genbank中人类基因的标准系列进行比对,测序识别的PITX2新突变与100位无亲缘关系的正常人进行测序对比分析。
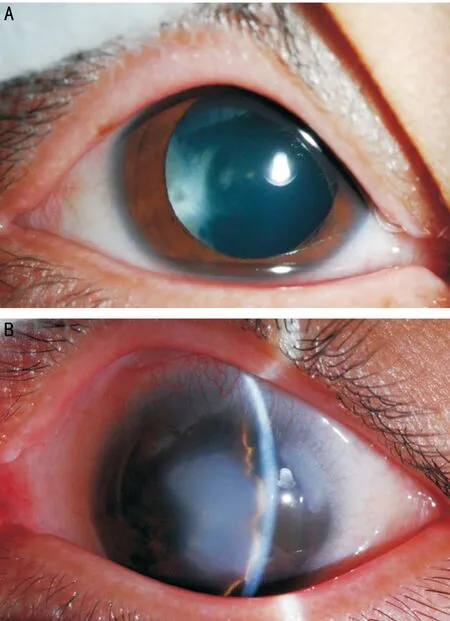
图1 突变患者双眼眼前节照相 A:右眼;B:左眼。
1.2.3 PITX2蛋白的生物信息学分析 从ensembl数据库中下载人类PITX2分子的氨基酸序列,采用在线工具(http://www.sbg.bio.ic.ac.uk/)中的Phyre2软件分析突变前后的PITX2蛋白质的三级结构。对突变位点可能导致的氨基酸变化的差异以及可能致病性通过PolyPhen软件进行评估。
2结果
在选取的10例先天性角膜混浊患者中,检测到1个新PITX2基因突变,分析其详细的相关临床资料,符合Peters综合征的临床诊断,这是中国首次报道Peters综合征的PITX2基因突变。
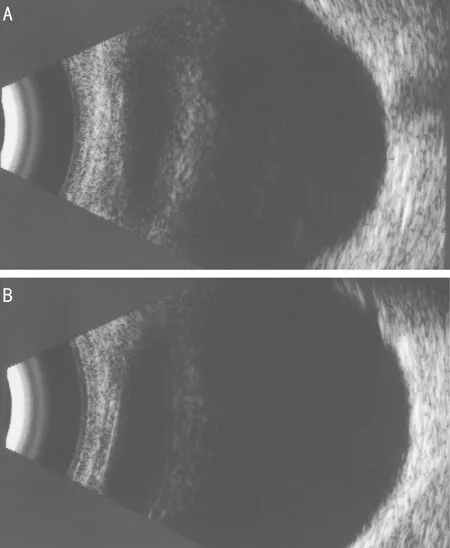
2.1 PITX2突变患者病例资料 患者,男,8岁,因双眼胀痛伴视物模糊1mo就诊。既往史:患者父母诉患儿出生后即被发现左眼角膜发白(患者双眼眼前节照相见图1)。4岁因患 “室间隔缺损”行室间隔缺损修补手术。7岁时被诊断出双耳听力下降。眼科检查:VOD:0.15(-1.00DS矫正0.2),VOS:0.1。右眼:角膜透明,前房深度可,晶状体白色混浊,眼底窥不清;左眼角膜中央变薄、白色圆形混浊大小约5mm,后弹力层变薄,前房轴深2CT,房角4∶00~6∶00角膜缘后可见后胚胎环,瞳孔圆,对光反射正常,散瞳后见晶状体透明,眼底检查窥不清;患儿A超检查双眼眼图2 突变患者眼部B超:双眼后节(-) A:右眼;B:左眼。
轴:OD:21.02mm,OS:21.00mm;双眼眼部B超:双眼后节(-)(图2)。房角镜检查:双眼房角粘连大于1/2,左眼可见Schwalbe线。辅助检查:NCT:OD 33mmHg,OS 43mmHg,给予盐酸卡替洛尔滴眼液、布林佐胺滴眼液点眼后,眼压:右眼12mmHg,左眼17mmHg。临床诊断:右眼白内障,左眼Peters综合征,双眼继发性青光眼,双耳听力损害,室间隔修补术后。
2.2基因突变筛查 基因突变筛查结果显示,该患者c.788G>A位点突变,而该基因的其他编码序列及外显子与内含子的交界处均未发现序列异常。该突变位于第五外显子内,编码区的第788个碱基由G突变成A,直接导致了其编码蛋白的改变,致使原蛋白的第69位氨基酸由精氨酸变成了组氨酸,正常及突变患者测序图对照见图3。该患儿家属中的正常个体及无亲缘关系的100位正常对照测序分析均未发现同样基因的突变,文献检索未见PPS病基因突变位点的报道。
2.3 PITX2蛋白的生物信息学分析 本研究中我们在1例PPS患者中发现一新的PITX2基因突变,该突变为杂合突变,编码区的第788个G碱基突变成A,对c.788G>A突变进行分析显示,该位点碱基的突变位于第五外显子内,直接导致了其编码蛋白的改变,致使原蛋白的第69位氨基酸由精氨酸变成了组氨酸。通过PolyPhen软件对该错义突变导致氨基酸变化的可能致病性进行分析,结果显示该变与致病性之间具有一定的关联性(软件分析结果:benign,0.003)。进一步用Phyre2软件构建c.788G>A突变前后的蛋白分子三级结构模型(图4)。
3讨论
3.1 Peters综合征临床特点 PPS是先天性角膜混浊眼病较为典型的类型,多见于婴幼儿或青少年期,其主要表现:先天性角膜中央部混浊白斑,伴有相应区域的角膜后部基质变薄和后弹力层缺损,虹膜角膜粘连,前房较浅,周边部角膜厚度可正常,角膜缘后可见后胚胎环,青光眼发病率在50%,小眼球伴发率达40%[1],另外可有眼部非特征性异常如前葡萄肿、巩膜化角膜、虹膜缺损及原始玻璃体增生症等表现。大多数是散发病例,但也有一些常染色体显性及隐性遗传的报道。该病通常被认为是一种单独的眼部疾病,但实质上35%患者可伴发全身发育的异常[2],如并发率在15%左右的生长发育迟缓、唇裂、心脏室间隔缺损、耳畸形、听力损害、中枢神经或泌尿生殖系统异常等。本研究中的该突变患者病例临床特点:(1)先天起病;(2)角膜中央变薄,白色混浊,后弹力层缺损;(3)虹膜角膜粘连,角膜缘后可见后胚胎环;(4)继发性青光眼;(5)同时伴有室间隔缺损以及听力损害、全身发育的异常。以上临床特点符合PPS的临床诊断[3]。目前PPS引起的青光眼可以采用药物或手术治疗,小梁切除术辅助抗代谢药物应用是首选。如角膜混浊局限,可长期散瞳治疗。如角膜混浊致密,应在出生后3mo内行穿透性角膜移植术,如果合并严重白内障,可同时行白内障手术以防止形觉剥夺性弱视。本例突变患儿,双眼青光眼予以盐酸卡替洛尔滴眼液、布林佐胺滴眼液点眼后眼压控制良好,患儿家属考虑予以药物保守治疗;右眼白内障则行白内障超声乳化及人工晶状体植入术,左眼Peters综合征引起的角膜混浊行穿透性角膜移植术,目前观察患儿双眼术后恢复良好。
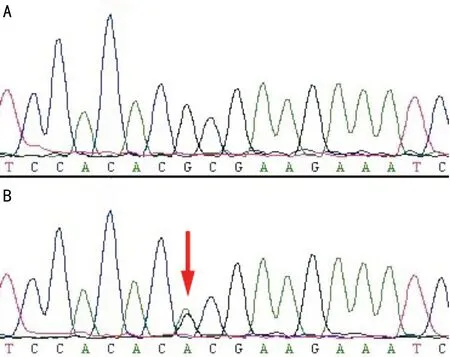
图3 正常及突变患者测序图 A:正常;B:突变。
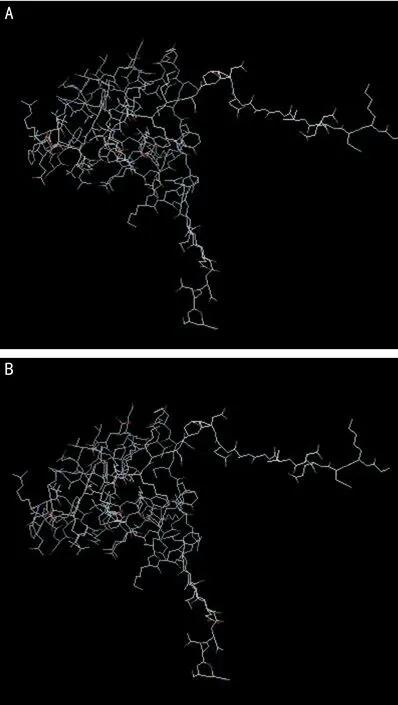
图4 PITX2野生型及突变型蛋白质分子三级结构模型 A:野生型;B:突变型。
3.2 PITX2基因功能和突变与Peters综合征发病机制 PITX2属于Paired-Bicoied同型盒蛋白家族,并与PITX1和PITX3构成PTX亚家族,定位于4q25-q27,全长19 930bp,形成271aa、317aa和324aa三种蛋白质亚型。这三种亚型主要区别在于氨基端的不同,而同源结构域以及羧基端的序列则完全相同。其包含一个60氨基酸同源结构域2(homeobox-2)和一个14氨基酸构成OAR(otp aristaless and rax)结构域,前者可通过其内在的“螺旋-旋转-螺旋”(helix-turn-helix,HTH)结构与特异性的DNA位点相结合,在个体发育过程中间发挥着重要作用,该同源结构域第50位氨基酸恒定为赖氨酸,对于特异性结合DNA具有决定性。后者推测与转录激活、蛋白质结合DNA有关。Peters综合征国外报道在新生儿中的发病率在1.2/100 000,致病基因定位在11P13区域PAX6基因和4q25区域PITX2基因,目前国外仅有较少Peter异常由PITX2基因突变致病的报道。1999年英国的Doward首次报道PITX2基因突变导致Peters综合征后,日本报道了1例Peters综合征患者的PITX2的突变[4];在国内,Peters综合征未见任何PITX2基因突变的报道。在本研究中,我们对10例先天性角膜混浊的患者进行PITX2基因突变的筛查,1例患者检测到一新的基因突变c.788G>A,临床特点及PITX2基因突变的研究均符合PPS的临床诊断, 这是中国首次报道PPS PITX2的基因突变。该碱基突变位于Exon5内,编码区的第788个碱基由G突变成A,该位点碱基的突变直接导致了其编码蛋白的改变,致使原蛋白的第69位氨基酸由精氨酸变成了组氨酸。推测PITX2基因的突变是PITX2蛋白结合不同靶基因DNA的关键区域,因此该位置突变将引起两者结合的不稳定性,从而影响蛋白质的功能。
3.3 Peters综合征基因型与表型的研究 PPS与Axenfeld-Rieger综合征(简称ARS)是两种不同类型的眼前段发育不良疾病,最新的研究证实胚胎学形成机制均是由神经嵴细胞的间充质细胞迁移分化异常引起。转录因子PITX2及FOXC1是这两种眼病的共同致病基因,基因的突变使神经嵴细胞的迁移分化过程出现异常,则会对眼前节的组织结构产生广泛的影响,从而导致PPS和ARS等眼前段疾病的发生[5];神经嵴细胞在心脏、骨、颅软骨、牙、真皮的形成发育中如受到阻碍,便可引起患者室间隔缺损以及听力损害等全身异常。国外文献曾报道了同一家系中FOXC1同一突变位点突变引起PPS和ARS两种眼前段发育不良的临床表型[6];有意思的是,国外文献报道了PITX2基因的同一位点c.788G>A突变导致Axenfeld-Rieger综合征病例[7],而本研究中的测序结果发现的同样位点c.788G>A突变导致PPS临床表型,PITX2可引起两种不同眼前段发育不良ARS和PPS眼病,似乎意味着两者眼病发病原因间的共同联系。在眼前段胚胎学形成发育过程中,多种基因的信号转导通路相互联系,形成复杂的网络体系,一个基因功能的实现依赖于该基因与其它基因间的相互作用,这些基因通过相互协作来控制生物体重要的生命过程,由此我们推测两种不同眼病的发生是由等位基因的突变或是PITX2和传导通路中的其他基因如FOXC1在共同的信号传导中的相互影响而导致。
PPS是临床少见病,但患者可伴发极高的青光眼发病几率,是严重影响患者视力的致盲性眼病,而国内资料较少,国内眼科医师对PPS认识不足,临床中经常被误诊。本研究分析了Peters综合征的临床特点,对其致病基因PITX2进行突变检测,阐明其发病的分子遗传学病因,以了解其基因型与表型的关系。今后需要更大规模、更深入地研究Peters综合征的病因,以及病因与临床表型之间的关系,从而为该眼病的诊断预防和治疗起到更全面的指导作用。
1 Mayer UM. Peters Anomaly and Combination with Other Malformations-(Series of 16 Patients).OphthalmicPaediatrGenet1992;13(2):131-135
2 Heon E, Barsoumhomsy M, Cevrette L,etal. Peters Anomaly - the Spectrum of Associated Ocular and Systemic Malformations.OphthalmicPaediatrGenet1992;13(2):137-143
3 Ozeki H, Shirai S, Nozaki M,etal. Ocular and Systemic Features of Peters’ Anomaly.GraefesArchClinExpOphthalmol2000;238(10):833-839
4 Arikawa A, Yoshida S, Yoshikawa H,etal. Case of Novel Pitx2 Gene Mutation Associated with Peters’ Anomaly and Persistent Hyperplastic Primary Vitreous.Eye2010;24(2):391-393
5 Sowden JC. Molecular and Developmental Mechanisms of Anterior Segment Dysgenesis.Eye2007;21(10):1310-1318
6 Weisschuh N, Wolf C, Wissinger B,etal. A novel mutation in the FOXC1 gene in a family with Axenfeld-Rieger syndrome and Peters’anomaly.ClinGenet2008;74(5):476-480
7 Kulak SC, Kozlowski K, Semina EV,etal. Mutation in the Rieg1 Gene in Patients with Iridogoniodysgenesis Syndrome.HumMolGenet1998;7(7):1113-1117
Clinical and genetic research in Chinese families with Peters syndrome
Li-Qin Huang1, Guo-Hua Lu1, Yang Xie1, Ping-An Mao1, Xin-Cheng Sun1, Yong Meng2
Foundation item:Guidance Project of Changzhou Municipal Health Bureau (No.WZ201315)1Department of Ophthalmology, Changzhou No.2 People’s Hospital, Changzhou 213000, Jiangsu Province, China;2Department of Ophthalmology, Changzhou No.3 People’s Hospital, Changzhou 213000, Jiangsu Province, China
Yong Meng. Department of Ophthalmology, Changzhou No.3 people’s Hospital,Changzhou 213000, Jiangsu Province, China. 13861047902@163.com
•AIM: To research the clinical characteristics and identify the disease-causing gene mutation in Chinese patients with Peters syndrome. All these cases will be useful foundations for clinical diagnosis, medical therapy and pathogenesis.
Peters syndrome; congenital corneal opacities; PITX2; mutation
常州市卫生局指导性项目(No.WZ201315)
1(213000)中国江苏省常州市第二人民医院眼科;2(213000)中国江苏省常州市第三人民医院眼科
黄丽琴,主治医师,研究方向:眼遗传病。
孟永,主治医师,研究方向:眼底病.13861047902@163.com
2016-08-03
2016-11-08
:Huang LQ, Lu GH, Xie Y,etal. Clinical and genetic research in Chinese families with Peters syndrome.GuojiYankeZazhi(IntEyeSci) 2016;16(12):2237-2240
10.3980/j.issn.1672-5123.2016.12.16
Received:2016-08-03 Accepted:2016-11-08
猜你喜欢
杂志排行

国际眼科杂志的其它文章
- Vision related quality of life and daily visual functioning in patients undergoing pan-retinal photocoagulation for proliferative diabetic retinopathy
- 不同焦点人工晶状体植入治疗白内障患者术后视觉效果
- Easy removal of rust rings formed after metal foreign bodies in cornea
- 白内障及屈光手术源性散光的计算方法汇总
- 屈光参差性弱视治疗后期加入视功能训练的临床观察
- JAK2-STAT3基因多态性对真菌性角膜溃疡患者伏立康唑血药浓度的影响