Clinical trial perspective for adult and juvenile Huntington's disease using genetically-engineered mesenchymal stem cells
2016-12-02PeterDengAudreyTorrestKariPollockHeatherDahlenburgGeralynAnnettJanNoltaKyleFink
Peter Deng, Audrey Torrest, Kari Pollock, Heather Dahlenburg, Geralyn Annett, Jan A. Nolta, Kyle D. Fink
Stem Cell Program and Institute for Regenerative Cures, University of California Davis Health System, Sacramento, CA, USA
INVITED REVIEW
Clinical trial perspective for adult and juvenile Huntington's disease using genetically-engineered mesenchymal stem cells
Peter Deng, Audrey Torrest, Kari Pollock, Heather Dahlenburg, Geralyn Annett, Jan A. Nolta, Kyle D. Fink*
Stem Cell Program and Institute for Regenerative Cures, University of California Davis Health System, Sacramento, CA, USA
Progress to date from our group and others indicate that using genetically-engineered mesenchymal stem cells (MSC) to secrete brain-derived neurotrophic factor (BDNF) supports our plan to submit an Investigational New Drug application to the Food and Drug Administration for the future planned Phase 1 safety and tolerability trial of MSC/BDNF in patients with Huntington's disease (HD). There are also potential applications of this approach beyond HD. Our biological delivery system for BDNF sets the precedent for adult stem cell therapy in the brain and could potentially be modified for other neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS), spinocerebellar ataxia (SCA), Alzheimer's disease, and some forms of Parkinson's disease. The MSC/BDNF product could also be considered for studies of regeneration in traumatic brain injury, spinal cord and peripheral nerve injury. This work also provides a platform for our future gene editing studies, since we will again use MSCs to deliver the needed molecules into the central nervous system.
mesenchymal stem cells; neurodegenerative disorders, Huntington's disease; genetic engineering; brain derived neurotrophic factor
Introduction
Huntington's disease (HD) is an autosomal dominant disorder caused by an expanded polyglutamine tract that results in progressive degeneration of neurons, primarily in the putamen, caudate nucleus and cerebral cortex. HD occurs when the gene that encodes huntingtin (Htt), located on the short arm of chromosome 4, shows an expanded CAG repeat region following exon 1 (The Huntington's Collaborative Research Group, 1993). Typically, greater than 38 CAG repeats correlates with onset of HD in mid-adulthood whereas a mutation greater than 60 CAG repeats results in an early-onset form of the disease known as juvenile Huntington's disease (JHD). Disease symptomology is clinically diagnosed by chorea, but early cognitive and emotional aberrations such as slowing of psychomotor speed, impairment of attention and memory, as well as executive and visuospatial functions that eventually degrade into dementia along with depression are becoming clearer in HD (Ross et al., 2014). JHD patients generally show less chorea than adult onset HD with rigidity and dystonia reported as the dominant clinical manifestations. Neuropathologically, HD is characterized by striatal atrophy, cortical thinning and degeneration in the prefrontal cortex with JHD also showing aberrations in the cerebellum (Aylward, 2007). Life expectancy is 15—20 years following clinical diagnosis in adult HD and 10 years in JHD (Gonzalez-Alegre and Afifi, 2006).
Pharmacotherapy for HD patients has been difficult despite increased recognition of the disorder, better access to genetic counseling and more availability to specialized care programs. Currently only palliative therapies that treat chorea (tetrabenzaine) and depression (SSRIs) are approved for HD patients. Furthermore, treatment for JHD has been incredibly challenging due to the complex symptomology and needs of pediatric patients. There are no studies to guide the current trial-and-error approach to treating JHD, with anti-parkinsonian agents, anti-psychotics and anti-epileptics being the mostly commonly prescribed drugs (Robertson et al., 2012).
It has been reported that HD patients have lower levels of brain derived neurotrophic factor (BDNF) due to inhibition at the transcriptional level by the mutant huntingtin protein (Zuccato et al., 2011). This reduction in BDNF in the striatum correlates with symptom onset and heightened severity of the disease in transgenic HD mice. Canonically, BDNF is known to mediate both the survival and function of striatal neurons. BDNF knockout mice recapitulate the striatal atrophy phenotype of HD patients and indicate that reduced neurotrophic support in the striatum is a major factor contributing to neurodegeneration in HD (Ciammola et al., 2007). Whereas, restoration of BDNF levels are shownto have pro-survival effects and ameliorate HD symptoms in transgenic HD rodent models. Therefore, BDNF is considered a prime candidate to treat the underlying neuronal dysfunction observed in HD (reviewed in Fink et al., 2015).
However, direct injections of BDNF have been ineffective due to the short half-life of the recombinant protein. Other exogenous delivery methods have been examined such as the use of adeno-associated viral (AAV) to express BDNF in striatal neurons. This has been shown to induce neurogenesis and promote longer life span in a HD mouse model. Interestingly, this effect was potentiated by noggin (Benraiss et al., 2012), which is a factor secreted by MSCs (Diefenderfer et al., 2003; Chen et al., 2012). Despite this success, the use of AAV in the clinic has proven difficult due to host immunogenicity to the virus and limited biodistribution (Kordower, 2016). In comparison, MSCs demonstrate paracrine effects such as local immune modulation and release of beneficial factors.
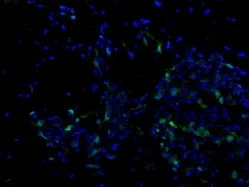
Figure 1 A representative image of transplanted human bone marrow mesenchymal stem cell that have been engineered with a green fluorescent protein reporter gene following transplantation into the brain of a transgenic Huntington's disease mouse model.
BDNF is initially synthesized by cortical neurons and then is transneuronally propagated into the striatum. When bound to TrkB, mature BDNF induces receptor dimerization and autophosphorylates tyrosine residues that initiates secondary cascades that regulate neurogenesis, synaptic plasticity (Huang and Reichardt, 2003). This is achieved by three major signaling pathways: the phosphatidyl inositol 3-kinase (PI3K)-serine-threonine kinase (AKT), mitogen-activated protein kinase (MAPK) extracellular related kinase (ERK) pathway, and the phospholipase Cy —Cam Kinase pathway.
Mesenchymal Stem Cells for HD
The use of stem cell therapies has become increasingly attractive as a putative therapy for neurodegenerative disorders. The ability to work synergistically with the endogenous microenvironment to upregulate intrinsic cell proliferation or neuroprotection via trophic factor secretion potentially enhances the overall regenerative potential in the transplanted tissue. Mesenchymal stem cells (MSCs) in particular have generated great interest in regenerative medicine and immunotherapy due to their unique biological properties. MSCs are multipotent stem cells derived from a broad subset of adult tissue that are readily accessible. MSCs are capable of secreting neurotrophic factors in response to local inflammatory elements, enhancing neurogenesis and synaptogenesis, and inhibiting apoptotic signaling. Due to their immune-modulating potential, MSCs do not require immunosuppression following allogenic transplantation and have demonstrated a strong safety profile in clinical trials (Figure 1).
A multitude of published articles have demonstrated improvement of either behavioral or neuropathological deficits in rodent models of HD following treatment with MSCs. These studies have used MSCs from multiple sources including autologous transplantation of unpurified whole bone marrow from rats, purified rat MSCs, mouse bone marrow-derived MSCs, mouse umbilical cord-derived MSCs, human adipose derived MSCs, and human bone marrow MSCs. Decreases in striatal atrophy, reduced medium spiny neuron loss, stimulation of endogenous neurogenesis andreduction of Htt aggregation has been observed following transplantation of MSCs (reviewed in Fink et al., 2015). Furthermore, MSCs have demonstrated improvements in motor and cognitive function, reduced anxiety-like behaviors and extension of lifespan in rodents.
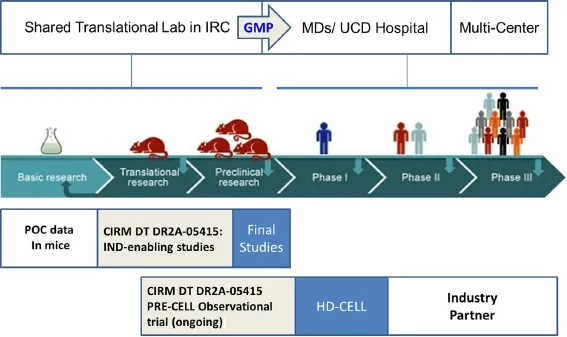
Figure 2 Investigational new drug (IND) trial design.
Genetically-Engineered Stem Cells
Trophic support is commonly postulated as the mechanism for the therapeutic potential of MSCs such as the secretion of neurotrophic factors that enhance endogenous neurogenic potential. Furthermore, the potential for MSCs as a delivery vehicle for gene therapy has been examined due to the relative ease of reprogramming these cells (Meyerrose et al., 2010). In particular, mouse-derived MSCs that were reprogrammed to overexpress BDNF in YAC128 HD transgenic mice resulted in motor improvement and increased neuron viability following transplantation (Dey et al., 2010). In addition to being readily programmable, MSCs migrate to areas of tissue damage unlike direct injection of AAV and demonstrate transient engraftment into host tissue as well as persistent sustained therapeutic effect even after cell clearance. In turn, this addresses safety concerns of direct use of viral vectors and associated prolonged immune response while still maintaining efficacy. Together, the use of MSCs to deliver BDNF as a treatment for HD has been suggested as a clinical therapy that our group is investigating in a series of Investigational New Drug (IND)-enabling preclinical studies. However, a controversial link between BDNF and epilepsy has been described in the literature, suggesting that sustained chronic overexpression may lead to deleterious effects. The evidence for the link between BDNF and epilepsy is due to the findings of increased levels of the trophic factor following limbic seizures, but conditional knockout or overexpression of BDNF does not severely alter kindling arguing against BDNF having a critical role in initiating seizure-like activity (Scharfman, 2005).
Summary of Current Work
Our group at the Institute for Regenerative Cures at the University of California, Davis has tested the safety and efficacy of genetically-engineered human bone marrow MSCs intransgenic HD mouse models and published the results of our IND-enabling studies in Molecular Therapy (Pollock et al., 2016). Human MSCs were isolated and cultured from commercially purchased whole bone marrow and transduced with a lentivirus designed to produce BDNF following Good Laboratory Practice-like conditions. These cells were prepared in a similar manner to cells proposed for a Phase I clinical trial. The YAC128 and R6/2 transgenic HD mouse models were used for behavioural and histological studies following transplantation to measure efficacy of MSC/BDNF as a putative therapy. Each mouse received intrastriatal transplantations of 5 × 105cells per hemisphere of untransduced MSC or MSC/BDNF with normosol-treated transgenic and wildtype littermate controls.
The ability of MSC/BDNF to reduce neuropathological deficits was measured by striatal volume 6 weeks post transplantation. It was observed that the YAC128 had approximately 11% striatal atrophy compared to WT. Interestingly, MSC alone reduced this striatal atrophy to approximately 8.4% and MSC/BDNF transplantations reduced it to approximately 6%. R6/2 treated with MSC/BDNF recapitulated the neurogenesis observed with non-modified MSCs, but also demonstrated an increase in mean survivability of 14.9% with MSC alone increasing lifespan 9.4% when compared to normosol treated transgenic mice. Finally, YAC128 mice treated with MSC/BDNF showed significantly less anxiety compared to normosol-treated YAC128 over a 6-week open field study. Taken all together, human MSC and MSC/ BDNF are both therapeutically relevant strategies in ameliorating HD symptomology observed in these two stains of transgenic mice. These results, along with the abundance of peer-reviewed articles (Fink et al., 2015) provide compelling evidence for the use of genetically-engineered MSC as a candidate therapy for changing the trajectory of disease progression in patients diagnosed with early-stage HD (Figure 2).
Next Steps (i.e., dosing, long term safety, large animal study, and application for an IND)
A host of rodent studies demonstrate the therapeutic potential of MSCs for HD, but there are current limitations to how well these models recapitulate the disease in humans. R6/2 mice are widely used to model behavioural deficits and extend the lifespan of these animals; however therapies aimed at preventing neuronal loss would be unsuccessful due to the lack of neuropathology in this model. Conversely, studies aimed at the metabolic dysfunction or at the extending the lifespan of mice in the YAC128 or BACHD mouse models would be unsuccessful as these mice exhibit weight gain that is uncharacteristic of the human condition and have a normal lifespan compared to wild-type littermates. Currently, large genetic animal models for HD are being pursued to open new avenues in HD research (Jacobsen et al., 2010). More importantly, large animals may better represent major neuroanatomical structures relevant in the human HD brain that are missing in rodent models. This will allow for large animal safety studies to accurately access delivery of stem cells and to perform long-term toxicology studies.
It is likely that any clinical study where MSCs are transplanted into human patients will hinge on the demonstration that they are both safe and feasible in such a trial. Currently, long-term engraftments of MSCs have shown varying success. Immune-suppressed mice xenografted with human MSCs show a month long engraftment potential whereas allogeneic MSC engraftments in macaque monkeys have been more variable due to presentation of immunogenicity (Isakova et al., 2014). Macaques transplanted with allogeneic/opposite sex MSCs demonstrated weak immune response, with increased transplantation size resulting in reduced engraftment duration suggestive that matched or partially matched cells from allogeneic sources may result in longer MSC engraftment. While MSC clinical trial design should still undergo further refinement, countless clinical trials havenot shown a need for Human Leukocyte Antigen (HLA) matching or immune suppression for stable MSC engraftment in humans (Fibbe et al., 2013). However, the transient nature of MSCs may be beneficial as a potential safeguard against prolonged immune response.
Current MSC Clinical Trials for Neurological Indications
MSCs have been well tolerated in phase I/II clinical trials without adverse events in a variety of diseases with no tissue matching. Currently, Atherys, SanBio and Brain-Storm Therapeutics have all concluded Phase II clinical trials with MSCs with no reported adverse effects in either acute or chronic neurodegenerative insults such as ischemic stroke or amyotrophic lateral sclerosis (ALS). Importantly, Brain-Storm Cell Therapeutics concluded a phase I/IIa clinical trial in patients with amyotrophic lateral sclerosis using autologous MSCs induced to express neurotrophic factor (NurOwn) with mild and transient adverse effects reported. Strikingly, treated ALS patients demonstrated slowed disease progression following the conclusion of the Phase IIa trial with improvements in breathing and reduced motor decline compared to pre-treatment (Petrou et al., 2016).
Currently, studies are underway to evaluate MSC/BDNF in a dose dependent manner and additional long-term safety studies in preparing of an IND application to the FDA. The proposed future clinical trial (HD-CELL) is designed to demonstrate safety of intrastriatal injection transplantation of genetically-modified MSCs to treat HD in patients screened in an ongoing observation study (PRE-CELL: Clinicaltrials.gov identifier NCT01937923) at the University of California, Davis. Given the growing support for MSCs in clinical trials of various neurodegenerative diseases, preclinical and in vivo biosafety data using human MSC/BDNF, and the current lack of treatments available for HD, it is believed that genetically engineered MSC are a strong lead candidate for the treatment of Huntington's disease.
Aylward EH (2007) Change in MRI striatal volumes as a biomarker in preclinical Huntington's disease. Brain Res Bull 72:152-158.
Benraiss A, Bruel-Jungerman E, Lu G, Economides AN, Davidson B, Goldman SA (2012) Sustained induction of neuronal addition to the adult rat neostriatum by AAV4-delivered noggin and BDNF. Gene Ther 19:483-493.
Chen C, Uludağ H, Wang Z, Jiang H (2012) Noggin suppression decreases BMP-2-induced osteogenesis of human bone marrow-derived mesenchymal stem cells in vitro. J Cell Biochem 113:3672-3680.
Ciammola A, Sassone J, Cannella M, Calza S, Poletti B, Frati L, Squitieri F, Silani V (2007) Low brain-derived neurotrophic factor (BDNF) levels in serum of Huntington's disease patients. Am J Med Genet B Neuropsychiatr Genet 144B:574-577.
Dey ND, Bombard MC, Roland BP, Davidson S, Lu M, Rossignol J, Sandstrom MI, Skeel RL, Lescaudron L, Dunbar GL (2010) Genetically engineered mesenchymal stem cells reduce behavioral deficits in the YAC 128 mouse model of Huntington's disease. Behav Brain Res 214:193-200.
Diefenderfer DL, Osyczka AM, Reilly GC, Leboy PS (2003) BMP responsiveness in human mesenchymal stem cells. Connect Tissue Res 44 Suppl 1:305-131.
Fibbe WE, Dazzi F, LeBlanc K (2013) MSCs: science and trials. Nat Med 19:812-813.
Fink KD, Deng P, Torrest A, Stewart H, Pollock K, Gruenloh W, Annett G, Tempkin T, Wheelock V, Nolta JA (2015) Developing stem cell therapies for juvenile and adult-onset Huntington's disease. Regen Med 10:623-646.
Gonzalez-Alegre P, Afifi AK (2006) Clinical characteristics of childhood-onset (juvenile) Huntington disease: report of 12 patients and review of the literature. J Child Neurol 21:223-229.
Huang EJ, Reichardt LF (2003) Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 72:609-642.
Isakova IA, Lanclos C, Bruhn J, Kuroda MJ, Baker KC, Krishnappa V, Phinney DG (2014) Allo-reactivity of mesenchymal stem cells in rhesus macaques is dose and haplotype dependent and limits durable cell engraftment in vivo. PLoS One 9:e87238.
Jacobsen JC, Bawden CS, Rudiger SR, McLaughlan CJ, Reid SJ, Waldvogel HJ, MacDonald ME, Gusella JF, Walker SK, Kelly JM, Webb GC, Faull RL, Rees MI, Snell RG (2010) An ovine transgenic Huntington's disease model. Hum Mol Genet 19:1873-1882.
Kordower JH (2016) AAV2-neurturin for Parkinson's disease: what lessons have we learned? Methods Mol Biol 1382:485-490.
Meyerrose T, Olson S, Pontow S, Kalomoiris S, Jung Y, Annett G, Bauer G, Nolta JA (2010) Mesenchymal stem cells for the sustained in vivo delivery of bioactive factors. Adv Drug Deliv Rev 62:1167-1174.
Petrou P, Gothelf Y, Argov Z, Gotkine M, Levy YS, Kassis I, Vaknin-Dembinsky A, Ben-Hur T, Offen D, Abramsky O, Melamed E, Karussis D (2016) Safety and clinical effects of mesenchymal stem cells secreting neurotrophic factor transplantation in patients with amyotrophic lateral sclerosis: Results of phase 1/2 and 2a clinical trials. JAMA Neurol 73:337-344.
Pollock K, Dahlenburg H, Nelson H, Fink KD, Cary W, Hendrix K, Annett G, Torrest A, Deng P, Gutierrez J, Nacey C, Pepper K, Kalomoiris S, D Anderson J, McGee J, Gruenloh W, Fury B, Bauer G, Duffy A, Tempkin T, et al. (2016) Human mesenchymal stem cells genetically engineered to overexpress brain-derived neurotrophic factor improve outcomes in Huntington's disease mouse models. Mol Ther doi:10.1038/mt.2016.12.
Robertson L, Santini H, O'Donovan KL, Squitieri F, Barker RA, Rakowicz M, Landwehrmeyer GB, Quarrell O (2012) Current pharmacological management in juvenile Huntington's disease. PLoS Curr 4:RRN1304.
Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS, Reilmann R, Unschuld PG, Wexler A, Margolis RL, Tabrizi SJ (2014) Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 10:204-216.
Scharfman HE (2005) Brain-derived neurotrophic factor and epilepsy--a missing link? Epilepsy Curr 5:83-88.
The Huntington's Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 72:971-983.
Zuccato C, Marullo M, Vitali B, Tarditi A, Mariotti C, Valenza M, Lahiri N, Wild EJ, Sassone J, Ciammola A, Bachoud-Lèvi AC, Tabrizi SJ, Di Donato S, Cattaneo E (2011) Brain-derived neurotrophic factor in patients with Huntington's disease. PLoS One 6:e22966.
10.4103/1673-5374.182682 http://www.nrronline.org/
Funding: Support for this project was provided by a NIH NIGMS Predoctoral Fellowship T32GM099608 (Deng), NIH NRSA Postdoctoral Fellowship F32NS090722 (Fink), a NIH Director's transformative award 1R01GM099688 (Nolta), A Stewart's and Dake Family Gift (Fink), California Institute for Regenerative Medicine (CIRM) DR2-05415 (Wheelock/Nolta), and philanthropic donors from the HD community, including the Roberson family and Team KJ.
How to cite this article: Deng P, Torrest A, Pollock K, Dahlenburg H, Annett G, Nolta JA, Fink KD (2016) Clinical trial perspective for adult and juvenile Huntington's disease using genetically-engineered mesenchymal stem cells. Neural Regen Res 11(5)∶702-705.
*Correspondence to: Kyle D. Fink, Ph.D., kdfink@ucdavis.edu.
orcid: 0000-0002-0235-5038 (Kyle D. Fink)
Accepted: 2016-04-15
杂志排行

中国神经再生研究(英文版)的其它文章
- Self-assembling peptide nanofibrous hydrogel as a promising strategy in nerve repair after traumatic injury in the nervous system
- Recovery of injured fornical crura following neurosurgical operation of a brain tumor: a case report
- Possible application of apolipoprotein E-containing lipoproteins and polyunsaturated fatty acids in neural regeneration
- Antibody-based neuronal and axonal delivery vectors for targeted ligand delivery
- Alzheimer's disease: the silver tsunami of the 21stcentury
- Adenosine A2Areceptors in neuronal outgrowth: a target for nerve regeneration?