Zr(F3)与2-丁炔分子自旋禁阻反应C—C,C—H键活化机理的理论研究
2016-09-07王永成马盼盼王文雪
王永成,马盼盼,王文雪
(西北师范大学化学化工学院,甘肃兰州 730070)
Zr(F3)与2-丁炔分子自旋禁阻反应C—C,C—H键活化机理的理论研究
王永成,马盼盼,王文雪
(西北师范大学化学化工学院,甘肃兰州730070)
运用密度泛函理论(DFT)B3LYP方法研究了单重态和三重态势能面自旋禁阻反应Zr活化2-丁炔分子的C—C和C—H键的反应机理.通过自旋-轨道耦合的计算讨论了势能面交叉点和可能的自旋翻转过程.反应从基态三重态开始,在活化C—C键的反应过程中出现了自旋态的改变,使得过渡态3T4g的活化能垒从-3.58降到-10.60 kJ·mol-1. 在MECP4处,单重态和三重态间的自旋-轨道耦合常数为146.10 cm-1,反应发生有效的系间窜越,从三重态跃迁到单重态势能面,反应势垒有所下降.
密度泛函理论(DFT);最低能量交叉点(MECP);自旋-轨道耦合(SOC);系间窜越(ISC)
过渡金属参与的氧化加成反应M+R-R′→R-M-R′,特别是催化活化碳氢化合物的反应,会出现C—C和C—H键的竞争反应[1-7].气相中过渡金属离子催化小分子的化学反应已经被广泛的研究[4,5,8,9].尤其是过渡金属原子参与的碳氢化合物中C—C键、C—H键活化反应一直是过渡金属催化反应的研究热点[6-10].HinRichs等[11]用交叉分子束的方法研究了Zr与2-丁炔的反应,发现反应中有H2和CH4放出,并且提出了可能的反应过程:
(1)
在饱和碳氢化合物(乙烷和丙烷)的催化活化过程中,过渡金属原子插入C—C键的势垒比C—H键高40~80 kJ·mol-1,因此只有活化C—H键的产物,即氢气的脱去过程更有利[2-3].与其相同,Zr催化2-丁炔的过程中Ecoll=71 kJ·mol-1时,C—C键断裂的产率为12%.
Li等[12]在理论上详细研究了脱去CH4的反应,但以单一势能面的绝热反应为前提.因过渡金属离子含有未充满的d轨道,它们既可以提供电子,也接受来自配体的电子,因此有较高的催化活性.基态Zr原子因d轨道上仅有2个分占4d轨道自旋平行的电子,在催化反应过程中,反应体系很可能在不同自旋态势能面间发生系间窜越,使反应向能量更低的路径进行[13,14].
1 计算方法与理论背景
1.1几何构型的优化
密度泛函理论[15]已广泛用于过渡金属化学的理论计算,其计算结果的准确性也被人们认同[16].本文采用DFT中Becke’s三参数交换泛函(B3)和Lee-Yang-Parr(LYP)相关泛函的混合DFT/Hartree-Fock的B3LYP方法[17],对C,H采用TZVP[18]全电子基组,对Zr采用Stuttgart/Dresden的双极化ξ(SDD)ECP基组[19],Zr的28个核芯电子被有效核势(ECP)取代[20],原子的价基组(4s24p64d25s2)从(8s7p6d)收缩为(6s5p3d).对反应体系的单、三重态势能面上的所有反应物、中间体、过渡态和产物的几何构型进行全参数优化,得到了对应稳定结构的热力学物理量、零点矫正能和振动频率.用频率分析证实了各反应物、中间体和产物的能量为局部极小,各过渡态有唯一虚频.对势能面上的每一个鞍点进行内禀反应坐标(IRC)计算,证实了反应坐标从鞍点分别走向反应物和产物.
在自旋禁阻反应历程中,不同自旋态势能面可能会在某处发生交叉,在交叉点处反应体系由一势能面系间窜越(Intersystem crossing, ISC)到另一势能面,并沿着能量最低的路径进行下去.为了寻找反应历程中单、三重态势能面交叉点位置,本文根据Yoshizawa等[21]报道的内禀坐标单点垂直激发方法,估算出不同自旋态透热势能面(Diabatic surface)间的交叉点(Crossing point,CP),再用Harvey等[22]提出的寻找最低能量交叉点(Minimum energy crossing point,MECP)的方法,采用Crossing 2004程序包确定了交叉点对应的MECP.本文主要计算工作运用Gaussian 03程序[23]完成.
1.2自旋-轨道耦合计算
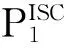
(2)
(3)
(4)
其中,HSOC为单、三自旋态之间自旋-轨道耦合矩阵元;ΔF为两不同自旋态势能面之间的梯度差;μ为MECP的折合质量;EMECP为MECP的相对能量.
2 结果与讨论
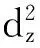
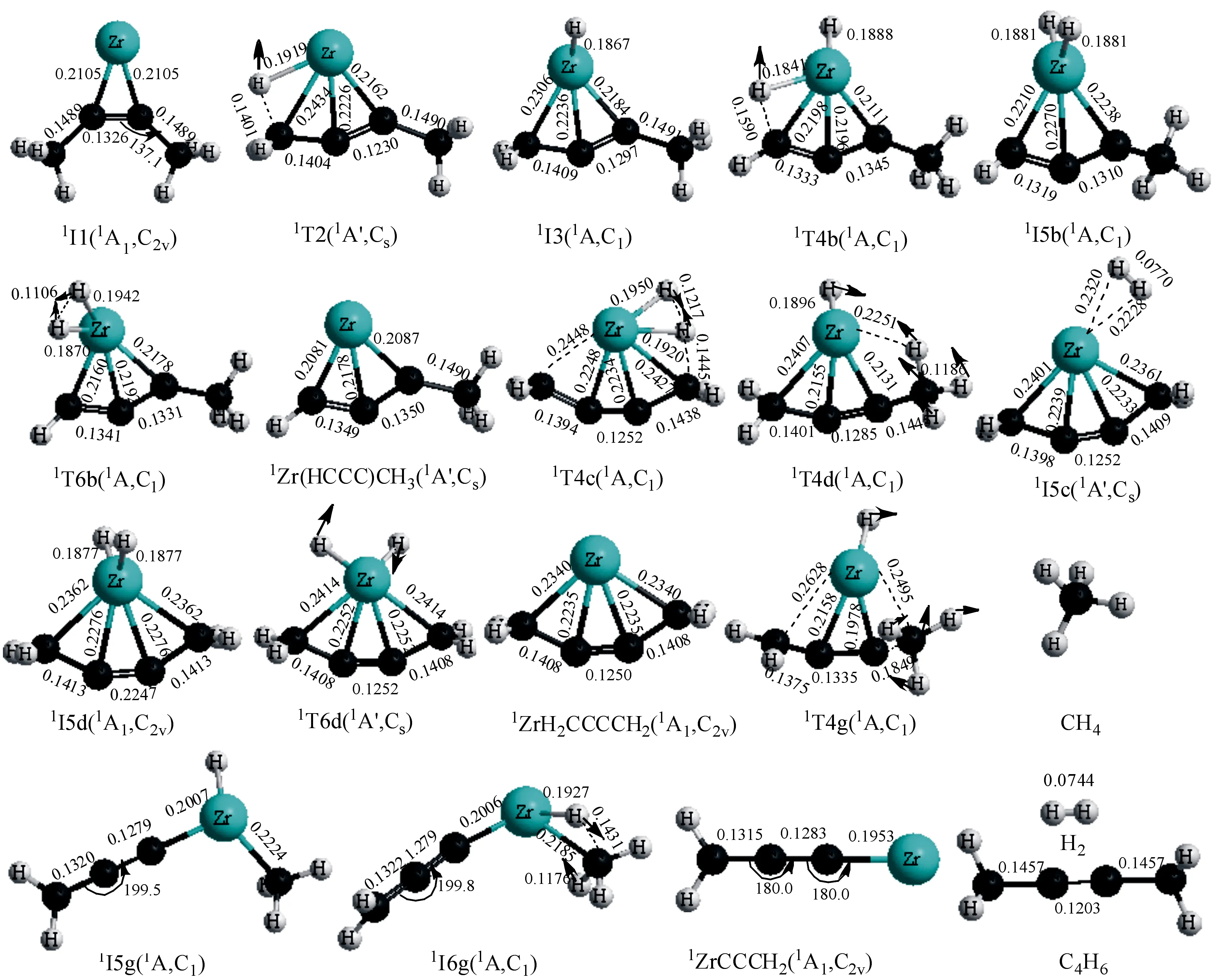
图1 在B3LYP/TZVP∪SDD水平下反应体系在单重态势能面上各驻点的几何构型(键长nm,键角(°))
2.1C—H键的活化
2.1.1同一甲基C—H键的活化如图2所示,a,b路径都是同一碳原子上H的离去反应,其中a,b路径分别是协同和分步脱氢过程.为了确定主要的反应通道,求得两种竞争路径的产物比例,根据图4 所示的3I3→3I5a和3I3→3I5b竞争反应用Gibbs自由能表示势能图,运用curtin-Hammett公式[30](5)可估算竞争反应的产物比例.
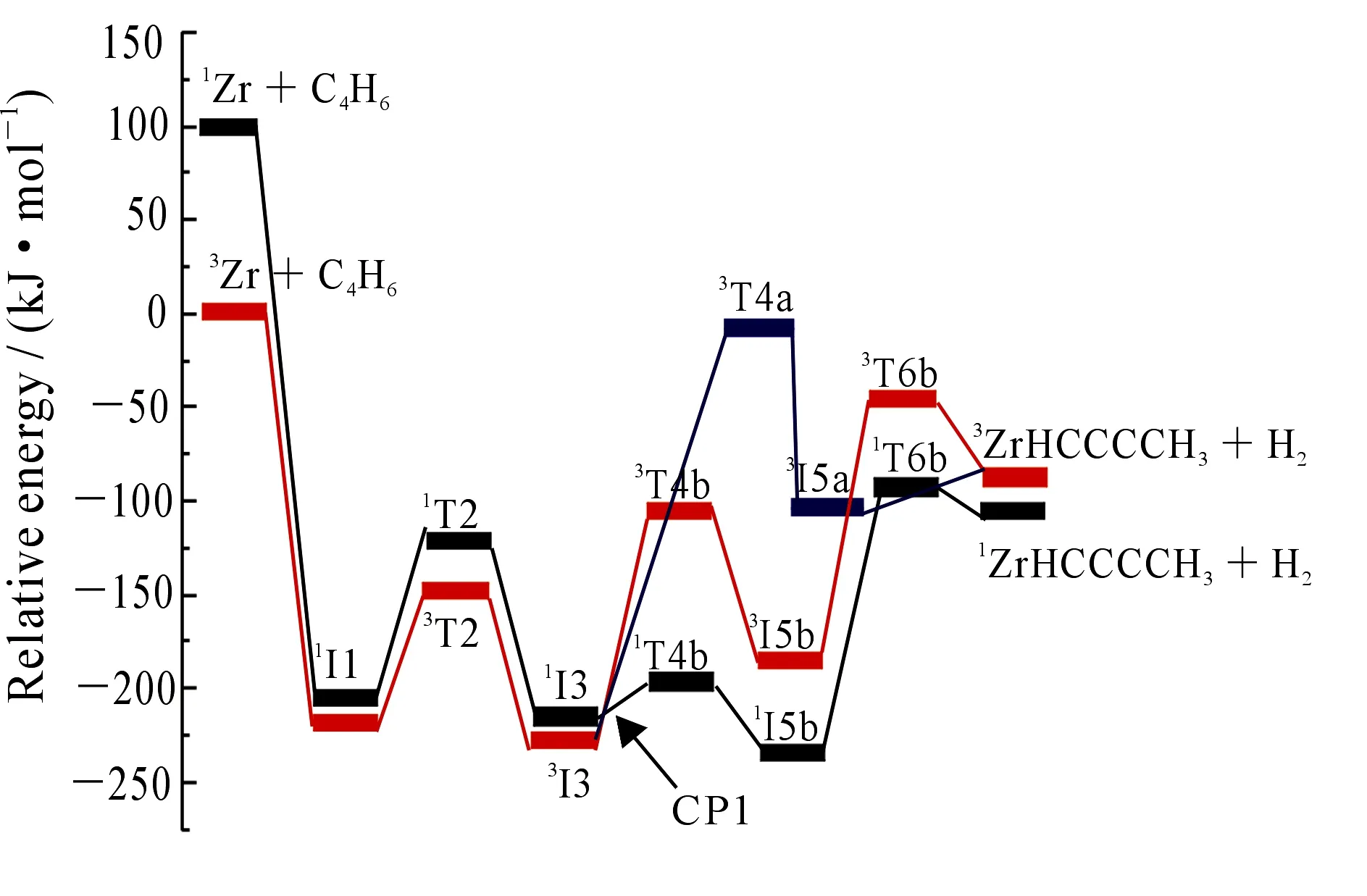
图2 B3LYP/TZVP∪SDD水平下a,b路径的势能面图
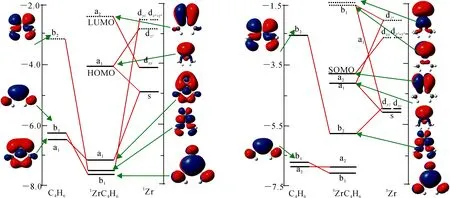
图3 单(a)、三(b)重态的复合物I1的轨道相互作用图
(5)
其中ΔΔG≠=ΔG≠(3T4b)-ΔG≠(3T4a).因过渡态3T4a比过渡态3T4b的Gibbs自由能高100.05kJ·mol-1,估算出竞争反应的产物比例接近于100%,可见在协同脱氢(路径a)和分步脱氢(路径b)的竞争反应中,路径b占据绝对优势.
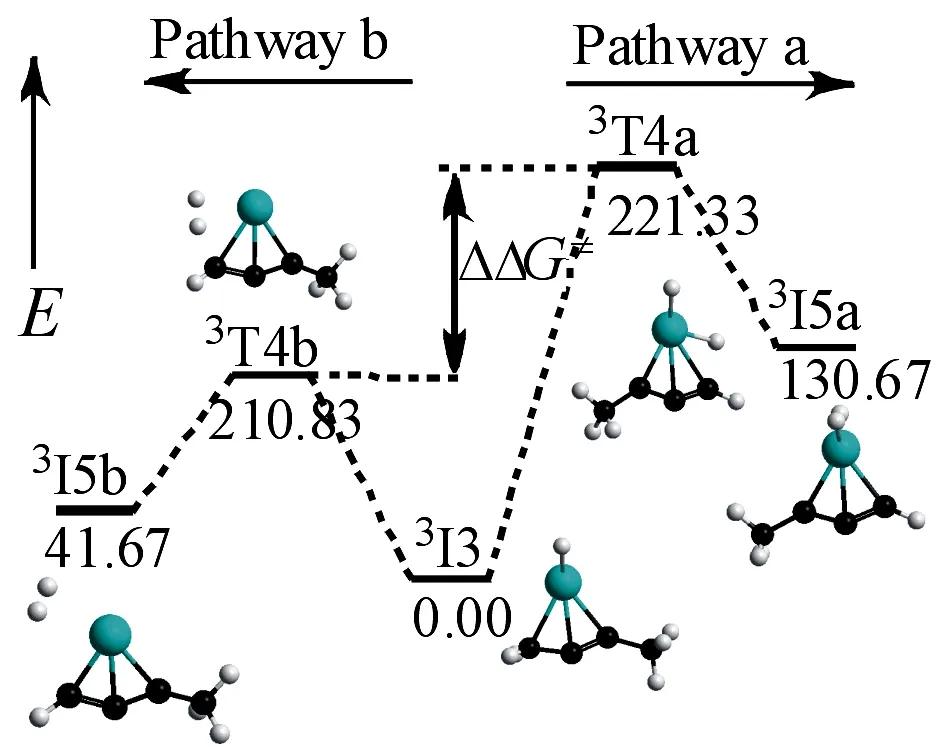
图4 3I3→3I5a/3I3→3I5b的相对吉布斯自由能图(kJ·mol-1)
2.1.2不同甲基C—H键的活化与a,b路径相同,如图5,c,d路径也分别是协同和逐步脱氢过程,不同点是c,d路径的2个氢原子是连接在不同甲基上的.经过渡态3T4c形成中间体3I5c需克服152.17 kJ·mol-1的能垒,3T4c的振动方向指向H2的离去.而形成中间体3I5d需跨越128.03 kJ·mol-1的能垒,3T4c的虚频为880.18i cm-1,其振动模式指向C—H键的断裂.较低的反应能垒说明d路径为主反应路径.为了进一步证明反应会朝向逐步脱氢过程,引入Curtin-Hammett原理,结果如图6,过渡态3T4d比过渡态3T4c的自由能低24.27 kJ·mol-1,相比较由过渡态3T4d生成的产物3T5d所占比例更高.
2.2C—C键的活化
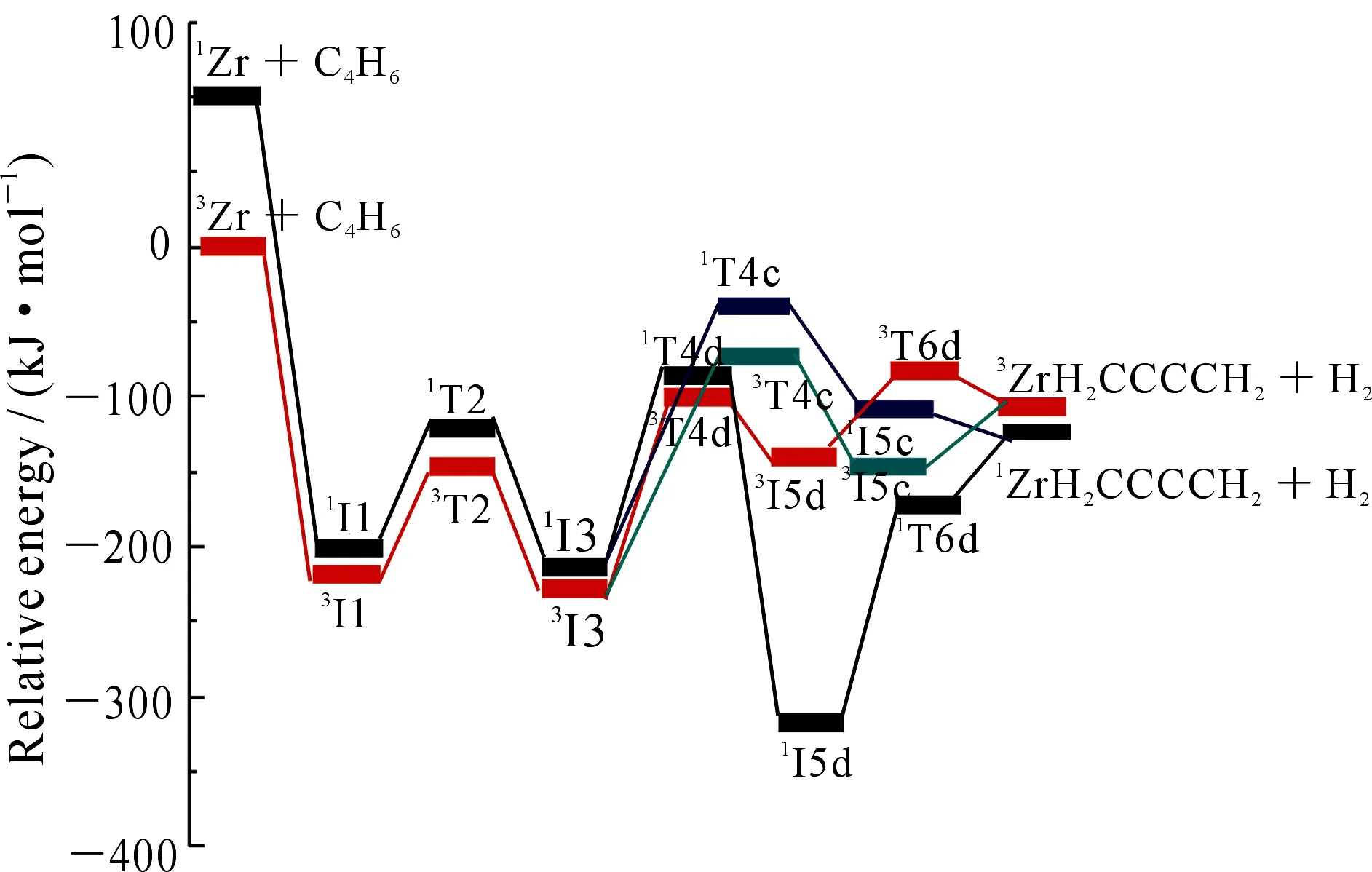
图5 B3LYP/TZVP∪SDD水平下c,d路径的势能面图
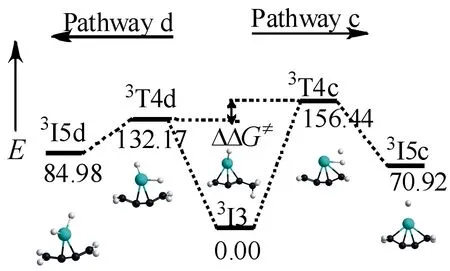
图6 3I3→3I5c/3I3→3I5d的相对吉布斯自由能图(kJ·mol-1)
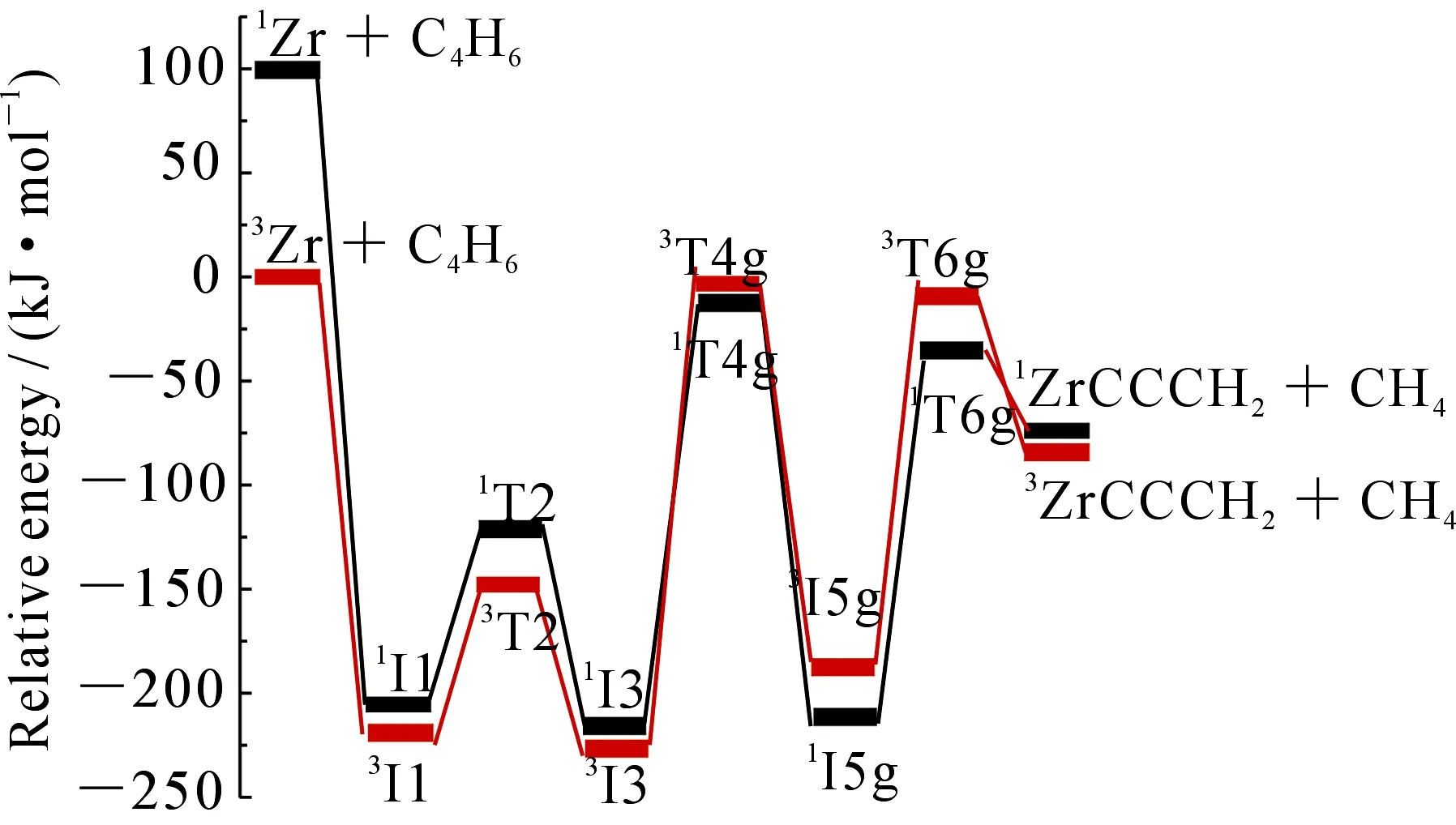
图7 B3LYP/TZVP∪SDD水平下g路径的势能面图
2.3势能面的交叉机理分析
从图2和图5可以看出,在脱去氢气的过程中反应以能量较低的基态三重态进入反应通道,以单重态离开反应通道,存在CP1、CP2和CP3三个交叉点.图7中生成CH4的路径有CP4和CP5两个交叉点,其中CP1,CP2,CP3和CP5位于过渡态之后,该交叉点在动力学上的作用不明显,只是决定了产物自旋态的分配,本文不做具体分析.如图7所示,从I3经过交叉区域到达T4g的过程中,两自旋态势能面较为接近,3T4g与1T4g之间的能差较小(约为7.02 kJ·mol-1),处于1/3I3与1/3T4g之间的交叉点CP4构型更接近于1I3,说明CP4处在中间体1I3附近.在CP4附近单、三重态发生混合,若在自旋-轨道耦合作用下发生有效的系间窜越,反应将会沿着能量更低的单重态势能面上进行,使得所翻越的势垒有所降低,相应的反应速率有所提高.
根据自旋-轨道耦合选律,ΔS=±1是自旋耦合作用发生的首要条件,CP4处的三重态向单重态跃迁时满足ΔS=-1.为了进一步探讨CP4附近非绝热过程行为,首先运用Harvey提出的能量梯度方法,运用Crossing 2004程序包优化了CP4对应的最低能量交叉点MECP4,对应的结构参数如图8所示.
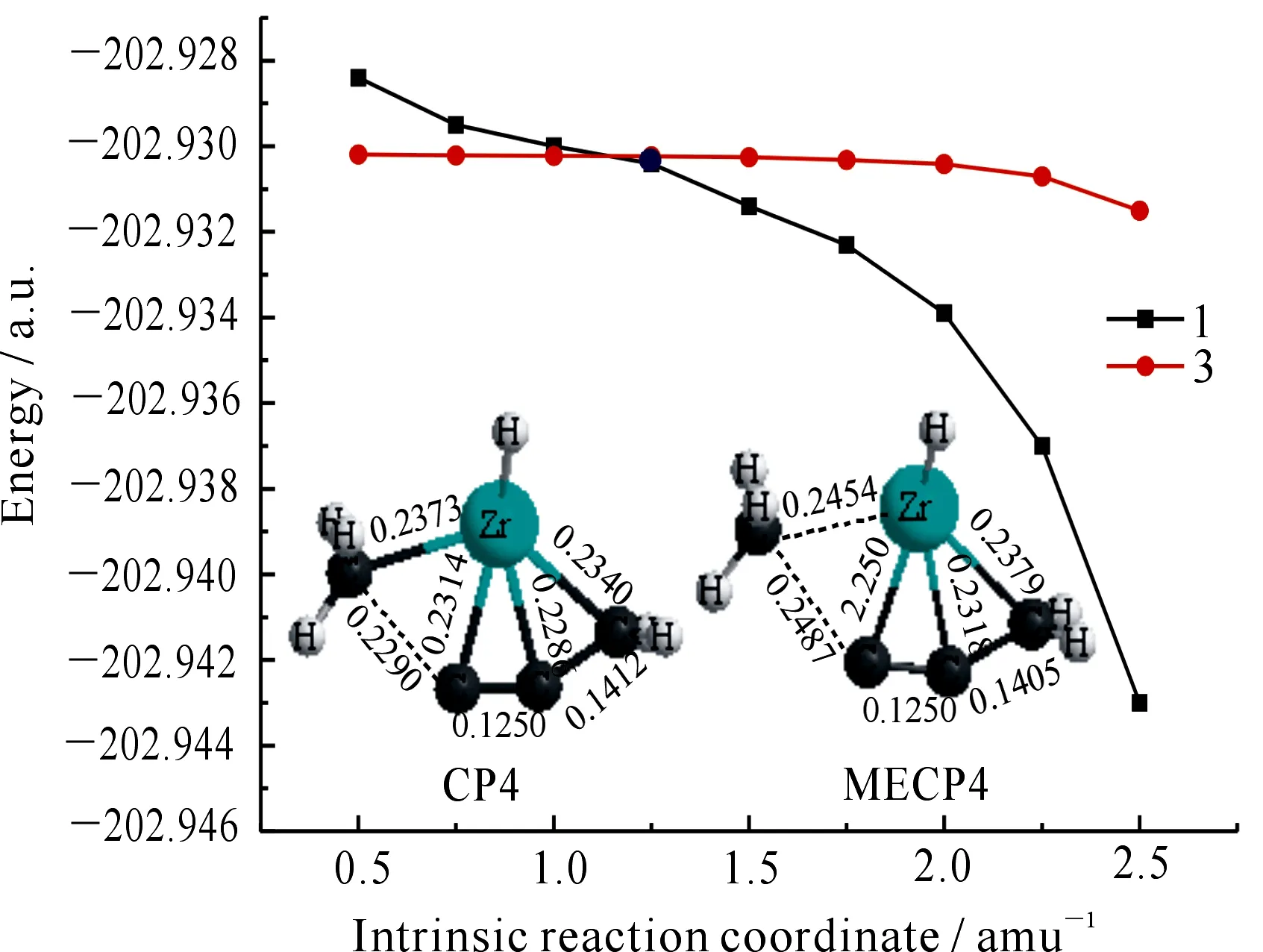
图8 交叉点CP4和最低能量交叉点MECP4的结构参数图
体系在MECP4处发生系间窜越时,电子的自旋态会发生改变,电子组态和前线分子轨道也随之改变.MECP4前线轨道能级图如图9,Ψa主要由Zr原子的dz2组成,它是三重态的最高单占轨道,也是单重态的最低空轨道;Ψb主要由Zr原子的dxz组成,它是三重态的最低单占轨道,也是单重态的最高占据轨道.得到d轨道矩阵元,即dz2ydxz.根据d轨道的关系得到y方向的角动量,在y算符的作用下,dz2和dxz允许改变其属性.在该步反应中,Ψa轨道上的α电子会翻为β电子到Ψb轨道上,完成三重态向单重态的转变.
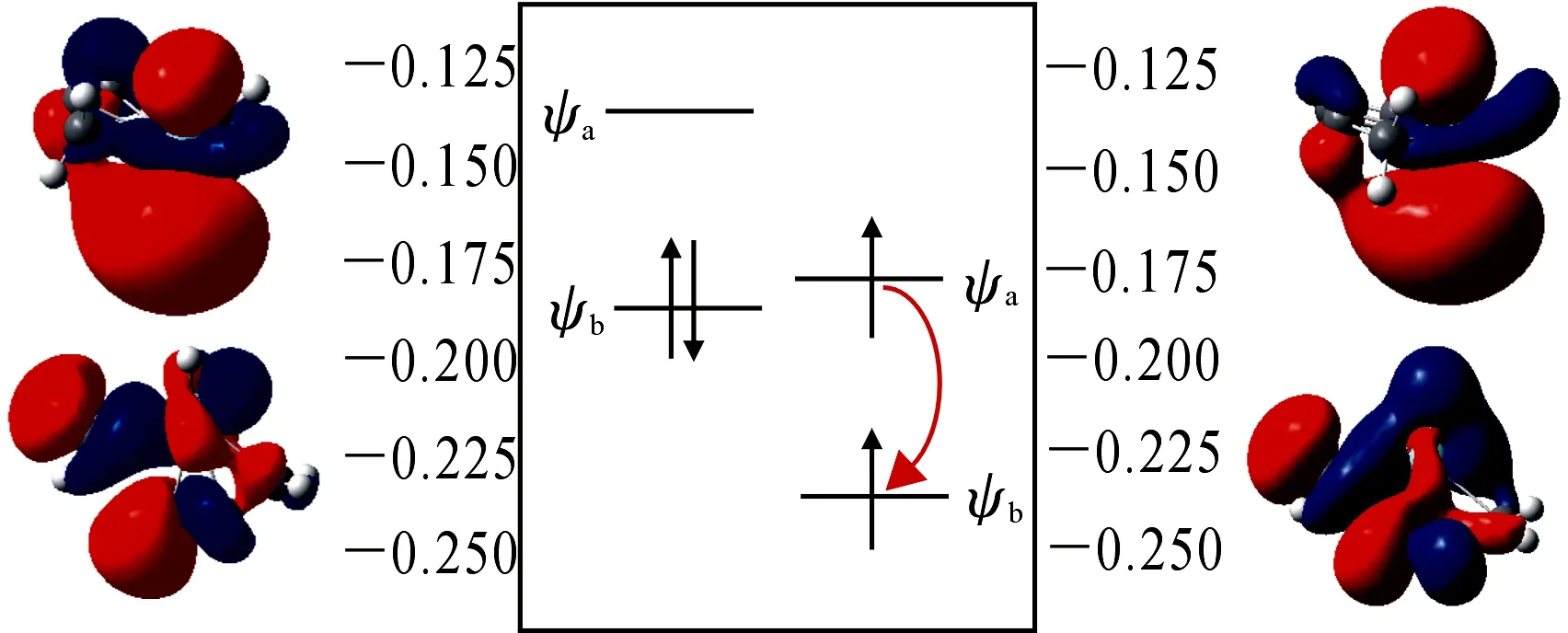
图9 前线分子轨道能级图
3 结论
在B3LYP/TZVP∪SDD理论水平下,对Zr催化活化2-丁炔脱去H2和CH4的两态反应做了计算研究,并运用Curtin-Hammett原理分析得逐步脱氢比协同脱氢过程更有利.在脱去甲烷的过程中,单重态和三重态在过渡态T4g前出现了势能面上的交叉点,较大的总系间窜越几率表明反应会沿着能量最低的路径进行,最后确定的最低能量反应路径为:
[1]SIEGBAHN P.The activation of the C-H bond in acetylene by second row transition metal atoms[J].Theoretica Chimica Acta,1994,87(4-5):277.
[2]CARROLL J J,HAUG K L,WEISSHAAR J C,et al.Gas phase reactions of second-row transition metal atoms with small hydrocarbons:Experiment and theory[J].The Journal of Physical Chemistry,1995,99(38):13955.
[3]STAUFFER H U,HINRICHS R Z,SCHRODEN J J,et al.Dynamics of H2and C2H4elimination in the Y+C2H6reaction[J].The Journal of Physical Chemistry A,2000,104(6):1107.
[4]ARMENTROUT P.Electronic state-specific transition metal ion chemistry [J].Annual Review of Physical Chemistry,1990,41(1):313.
[5]WEISSHAAR J C.Bare transition metal atoms in the gas phase:reactions of M,M+,and M2+with hydrocarbons[J].Accounts of Chemical Research,1993,26(4):213.
[6]SCHRODEN J J,WANG C C,DAVIS H F.Competition between C-C and C-H activation in reactions of neutral yttrium atoms with four butene isomers[J].The Journal of Physical Chemistry A,2003,107(44):9295.
[7]HINRICHS R Z,SCHRODEN J J,DAVIS H F.Competition between C-C and C-H insertion in prototype transition metal-hydrocarbon reactions[J].Journal of the American Chemical Society,2003,125(4):860.
[8]SCHRODEN J J,DAVIS H F.Reactions of neutral transition metal atoms with small molecules in the gas phase[C]//Modern Trends in Chemical Reaction Dynamics Series:Advanced Series in Physical Chemistry 14.Beijing:Science Press,2004:215.
[9]甘延珍,王永成,王环江,等.Fe+(6D)催化N2O和CH4制取甲醇循环反应的理论探究[J].中国科学:化学,2013,43(6):763.
[10]王永成,李雅婷,蔡君,等.Fe+催化N2O与H2反应的理论探究[J].西北师范大学学报(自然科学版),2014,50(3):71.
[11]HINRICHS R Z,SCHRODEN J J,DAVIS H F.C—C versus C—H bond activation of alkynes by early second-row transition metal atoms[J].The Journal of Physical Chemistry A,2008,112(14):3010.
[12]LI T H,WANG C M,XIE X G.Methyl and methane elimination in the gas phase reaction of zirconium atom with 2-butyne:a DFT study[J].Chinese Chemical Letters,2010,21(12):1501.
[13]HARVEY J N,POLI R,SMITH K M.Understanding the reactivity of transition metal complexes involving multiple spin states[J].Coordination Chemistry Reviews,2003,238:347.
[14]SCHRÖDER D,SHAIK S,SCHWARZ H.Two-state reactivity as a new concept in organometallic chemistry[J].Accounts of Chemical Rresearch,2000,33(3):139.
[15]SU M D,CHU S Y.Density functional study of some germylene insertion reactions[J].Journal of the American Chemical Society,1999,121(17):4229.
[16]HARVEY J N.On the accuracy of density functional theory in transition metal chemistry[J].Annual Reports Section,2006,102:203.
[17]LEE C,YANG W,PARR R G.Development of the colle-salvetti correlation-energy formula into a functional of the electron density[J].Physical Review B,1988,37(2):785.
[19]LEININGER T,NICKLASS A,STOLL H,et al.The accuracy of the pseudopotential approximation.Ⅱ.A comparison of various core sizes for indium pseudopotentials in calculations for spectroscopic constants of inH,inF,and inCl[J].The Journal of Chemical Physics,1996,105(3):1052.
[20]LUO Q,LI Q S,YU Z H,et al.Bonding of seven carbonyl groups to a single metal atom:theoretical study of M(Co)n(M=Ti,Zr,Hf;n=7,6,5,4)[J].Journal of the American Chemical Society,2008,130(24):7756.
[21]YOSHIZAWA K,SHIOTA Y,YAMABE T.Intrinsic reaction coordinate analysis of the conversion of methane to methanol by an iron-oxo species:a study of crossing seams of potential energy surfaces[J].The Journal of Chemical Physics,1999,111(2):538.
[22]HARVEY J N,ASCHI M,SCHWARZ H,et al.The singlet and triplet states of phenyl cation.A hybrid approach for locating minimum energy crossing points between non-interacting potential energy surfaces[J].Theoretical Chemistry Accounts,1998,99(2):95.
[23]KUDIN K,BURANT J,MILLAM J,et al.Gaussian 03,revision e.01.Gaussian Inc,2004.
[24]POLI R,HARVEY J N.Spin forbidden chemical reactions of transition metal compounds:new ideas and new computational challenges[J].Chemical Society Reviews,2003,32(1):1.
[25]GRANOVSKY A A.GAMESS:Version 7.0 [EB/OL].[2009-06-10].http://classic.chem.msu.su/gran/gamess/index.html.
[26]HARVEY J N,ASCHI M.Modelling spin-forbidden reactions:recombination of carbon monoxide with iron tetracarbonyl[J].Faraday Discussions,2003,124:129.
[27]HARVEY J N.Understanding the kinetics of spin-forbidden chemical reactions[J].Physical Chemistry Chemical Physics,2007,9(3):331.
[28]LU T,CHEN F.Multiwfn:A multifunctional wavefunction analyzer[J].Journal of Computational Chemistry,2012,33(5):580.
[29]DAPPRICH S,FRENKING G.Investigation of donor-acceptor interactions:a charge decomposition analysis using fragment molecular orbitals[J].The Journal of Physical Chemistry,1995,99(23):9352.
[30]SEEMAN J I.The Curtin-Hammett principle and the winstein-holness equation:new definition and recent extensions to classical concepts[J].Journal of Chemical Education,1986,63(1):42.
(责任编辑陆泉芳)
Theoretical study of mechanism for C—C,C—H bond activation in spin-forbidden reaction between Zr and 2-butyne
WANG Yong-cheng,MA Pan-pan,WANG Wen-xue
(College of Chemistry and Chemical Engineering,Northwest Normal University,Lanzhou 730070,Gansu,China)
The mechanism of the spin-forbidden reaction Zr(3F) and 2-butyne on singlet and triplet potential energy surface(PESs) has been investigated by DFT of B3LYP.Crossing points between the different potential energy surfaces and the possible spin inversion process are discussed by spin-orbit coupling(SOC) calculations.The reacting system starts in the triplet ground state,the changes of the spin state probably occur the activation of C—C,lead to a significant decrease in the barrier height of transition state3T4g from -3.58 to -10.60 kJ·mol-1.The values of the SOC constants at minimum energy crossing point(MECP4) are 146.10 cm-1between singlet and triplet PESs.The effective intersystem crossing(ISC) between different triplet and singlet PESs can occur and obviously reduce energy barriers.
density functional theory(DFT);minimum energy crossing point(MECP);spin-orbit coupling(SOC);intersystem crossing(ISC)
10.16783/j.cnki.nwnuz.2016.02.014
2015-09-26;修改稿收到日期:2015-11-18
国家自然科学基金资助项目(21263023)
王永成(1956—),男,陕西户县人,教授,博士研究生导师.主要研究方向为化学动力学.
E-mail:ycwang@163.com
O 643.1
A
1001-988Ⅹ(2016)02-0068-07
