氢氦在钨中的影响的第一性原理研究
2016-07-26欧阳晓平
张 峥,赵 强,*,欧阳晓平,2,3
1.华北电力大学 核科学与工程学院 非能动核能安全技术北京市重点实验室,北京 102206;2.西北核技术研究所,陕西 西安 710024;3.湘潭大学 材料科学与工程学院,湖南 湘潭 411105
氢氦在钨中的影响的第一性原理研究
张峥1,赵强1,*,欧阳晓平1,2,3
1.华北电力大学 核科学与工程学院 非能动核能安全技术北京市重点实验室,北京102206;2.西北核技术研究所,陕西 西安710024;3.湘潭大学 材料科学与工程学院,湖南 湘潭411105
摘要:采用PBE形式的广义梯度近似(GGA)的第一性原理计算方法研究了氢或氦在钨中产生点缺陷的形成能以及缺陷形成后对钨的弹性的影响;采用同样的方法研究了空位和自间隙原子这两种缺陷。经计算发现:氢氦掺杂在钨的晶体结构中会引起晶体体积的变化,其变化结果跟掺杂的位置有关,在四面体或八面体处的掺杂会使晶体体积增加,替位掺杂会引起晶体体积减小;从形成能来看,氢掺杂在钨中最占优的位置是四面体处,而氦最占优的则是替位掺杂。在几种缺陷中,形成能最小的是氢的四面体掺杂,形成能最大的则是钨的自间隙原子形成;钨中若含有氢或氦的点缺陷,晶体的体弹模量和剪切模量会发生改变,当钨中含有氢替位或自间隙原子时晶体会向塑性改变,含有其他点缺陷时晶体会沿着脆性方向转变。但总体来说带有缺陷的钨仍然具有延展性。值得注意的是,氢或氦在钨中会引起晶体的各向异性,其具体结果与缺陷所处位置相关,只有缺陷属于替位时才不会发生各向异性。本文的研究工作可为第一壁材料的开发提供理论参考。
关键词:氢;氦;钨;第一性原理
核聚变能是潜在的清洁安全能源[1]。核聚变作为21世纪最理想的能源也受到了越来越多的关注。特别是2006年以来,我国经过谈判加入了国际热核聚变实验堆(International Thermonuclear Experimental Reactor, ITER)项目[2-3],该项目是仅次于国际空间站的国际合作的第二大合作项目。聚变堆的第一壁是实验包层模块直接面向等离子体的部件,因此,其组成材料又被称为等离子体面壁材料[4](plasma facing materials, PFMs)。由于第一壁材料面对的是一种极端环境,所以寻找和研制第一壁材料成为ITER能否安全运行的关键问题之一。目前,人们已经尝试了多种可能的第一壁材料,其中最典型的第一壁候选材料是铍(Be)[5-10]、碳(C)基材料[10-14]和钨(W)及其合金[15-30]。钨及其合金具有低的溅射率和良好的热力学性质(比如耐高温、高熔点和优良的热传导性能)被普遍地认为是最具有潜力的候选材料。氢氦分别作为热核聚变反应的反应物和生成物,均具有很高的能量,能够作为杂质掺入钨中形成缺陷。聚变反应产生的中子的能量约为14 MeV,如此高能量的中子能够使钨中形成空位和间隙原子这两种点缺陷。第一性原理计算方法已经被大量地用来研究钨或钨中的缺陷,并且也取得了一定成果。如:王欣欣等[1]采用第一性原理研究了铌对钨中氦行为的影响,徐景城等[31]通过第一性原理方法研究了完美钨晶体中的氢,Abdullah等[32]使用第一性原理计算方法研究了氧在钨中的扩散与溶解。本工作主要是研究氢氦掺杂钨、钨中空位和间隙原子的形成能、以及缺陷形成后钨中弹性常数的变化,文中所有的研究工作均是基于第一性原理计算展开的。
1计算模型和方法
1.1计算模型
钨属于体心立方晶系,所属空间群符号为Im-3m,α=β=γ=90°,a=b=c=0.316 5 nm。其晶体结构示于图1,每一小图中都包含一个钨的2×2×2的超晶胞。氢氦在钨中的掺杂方式和位置有替位(substitutional site)掺杂和间隙(interstitial site)掺杂。而间隙掺杂又包括四面体(tetrahedral site)间隙掺杂和八面体(octahedral site)间隙掺杂,文中分别记为Hesub、Hetet、Heoct、Hsub、Htet、Hoct。作为第一壁材料,钨通常要经受高能聚变中子的辐照,容易在其中形成空位(vacancy)和间隙原子(interstitial atom),文中记为Wvac和Wint。
1.2计算方法
本工作的计算采用一种基于DFT(density functional theory)的从头算量子力学程序CASTEP(Cambridge sequential total energy package)。它利用平面波赝势方法,将离子势用赝势代替,电子波函数通过平面波基组展开,电子-电子相互作用的关联势由LDA(local density approximation)或GGA(generalized gradient approximation)进行描述。本工作中计算所用晶格参数均为实验值,计算中采用PBE(Perdew-Burke-Ernzerhof)形式[33]的GGA近似的超软赝势和BFGS(Broyden-Fletcher-Goldfard-Shanno)方法对所建立的晶体模型进行结构优化,再对它们的能量和弹性常数进行计算。本文计算中初始设置参数如下:平面波截断能量300 eV,自洽收敛精度为每原子5×10-7eV,原子间相互作用力收敛标准为0.01 eV/nm,单原子能量收敛标准为5×10-6eV,晶体内应力收敛标准为0.02 GPa,布里渊区的积分采取5×5×5的特殊K点对全布里渊区求和,能量计算都在倒易空间中进行。结构优化完成的标志是四个参数均达到或优于收敛标准,计算结果与实际情况符合良好。
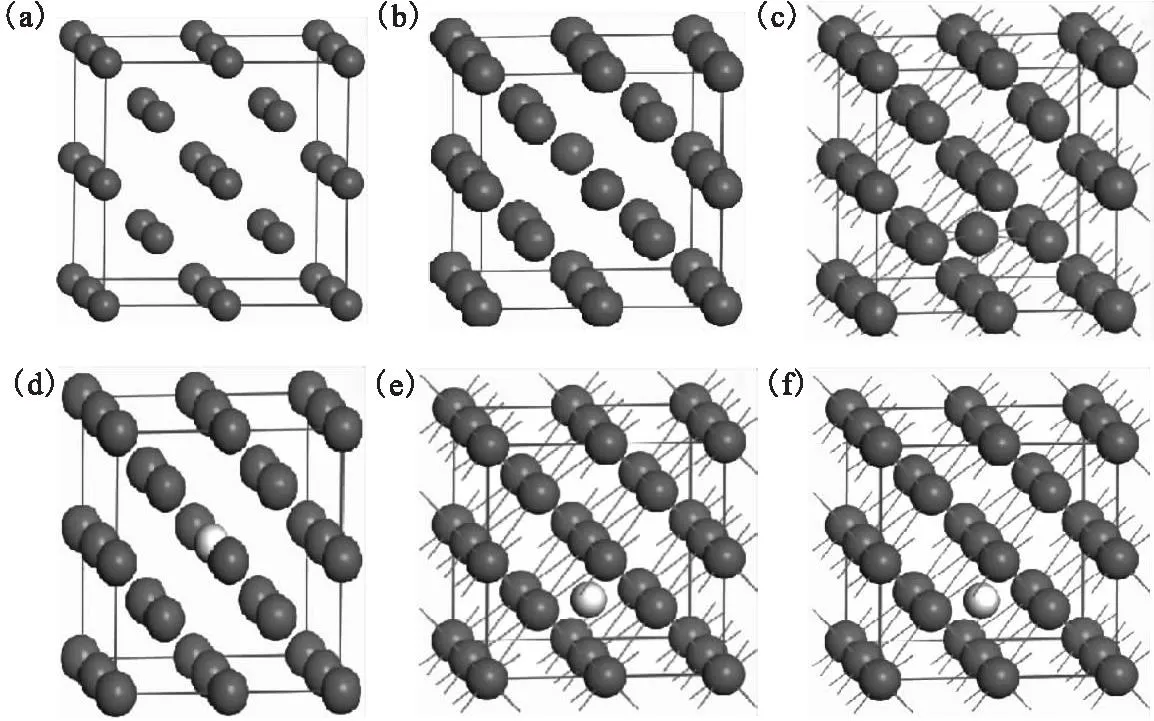
(a)——纯钨的超晶胞,(b)——钨空位,(c)——钨原子自间隙,(d)——替位取代,(e)——四面体位,(f)——八面体位深色小球为钨原子,浅色小球为掺杂原子图1 计算模型Fig.1 Computing models
2计算结果及讨论
2.1晶体结构优化
为了验证计算所选取的参数,通过对带有各种缺陷的钨的晶体进行几何结构优化,计算出的钨的晶体参数列于表1。从表1中可以看到钨的棱长为0.318 3 nm,这与实验值[34]0.316 5 nm符合良好。由于掺杂,原来的一个超晶胞变为一个新的晶胞,故棱长度大约变为原来的2倍。与纯钨相比,氦的八面体和四面体掺杂钨使晶胞体积有所增加,而氦的替位掺杂钨时晶胞体积减小。对于氢来说,当它掺杂位置处于四面体时晶胞体积增加,其余位置掺杂钨会使晶胞体积减小。当钨产生空位点缺陷时晶胞的体积减小,而含有自间隙钨原子时增加。从总体来看,处于四面体和八面体的点缺陷会引起晶胞体积增加,而氢氦替位掺杂会引起晶胞体积的减小。这些现象的产生是由于不同原子的半径各不相同,替位掺杂使晶胞体积减小,外来原子的间隙位置掺杂易引起晶胞体积增加。掺杂后晶胞体积的变化还与原子间的势场及键的变化相关。
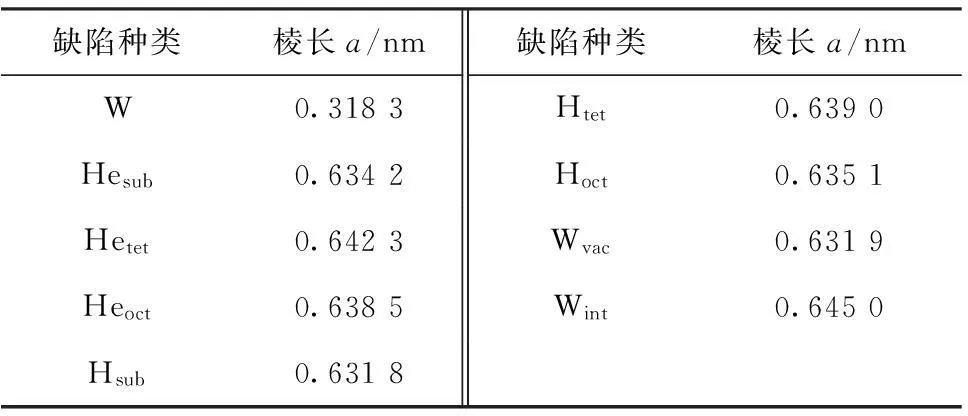
表1 钨及其缺陷物的晶胞常数
2.2缺陷的形成能
在晶体中形成一个缺陷(defect)需要一定的能量,通常把整个能量称为形成能。它的表达式如下:
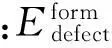
通过计算晶体中的缺陷形成能,能够判断哪种缺陷在晶体中更为稳定。表2是本工作计算得到的在钨中各种缺陷的形成能与其他学者研究结果[31,35]和实验结果[36]的对比。从表2可以看出,氦在钨中最容易形成的缺陷是替位掺杂,三种掺杂形式均为吸热反应。而氢在钨中最容易在四面体位掺杂,只有替位掺杂为吸热反应,其余为放热反应。空位的形成能比氦掺杂的形成能低,故形成空位应比氦掺杂更容易。由表2还可以看出,形成自间隙原子的能量相对于其他几种点缺陷的形成能大很多。本工作的计算结果与其他学者的研究结果符合良好。
表2钨中单一缺陷的形成能
Table 2Formation energy of the single defects in tungsten
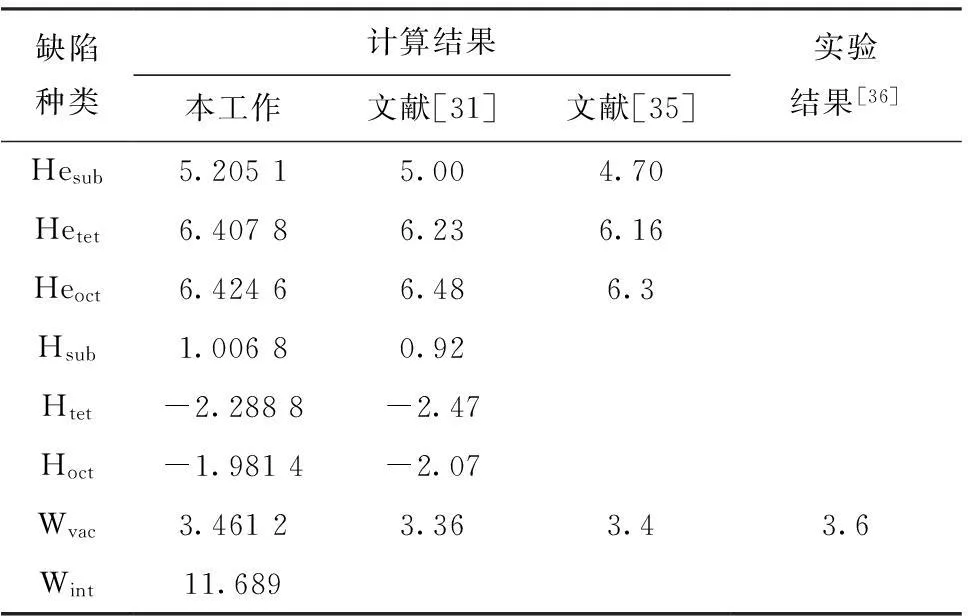
缺陷种类计算结果本工作文献[31]文献[35]实验结果[36]Hesub5.20515.004.70Hetet6.40786.236.16Heoct6.42466.486.3Hsub1.00680.92Htet-2.2888-2.47Hoct-1.9814-2.07Wvac3.46123.363.43.6Wint11.689
2.3弹性常数
弹性常数和体弹模量是表征材料的重要参数,反映了材料的稳定性和硬度。通过弹性常数可以得到与晶体的力学和动力学相关的信息。因此,获取准确的材料弹性常数十分必要。本工作计算了钨(及其含有缺陷)的弹性常数,计算结果列于表3和表4。从表3可以看到,本工作计算得到完美的钨的体弹模量为318.168 GPa,此结果与文献[36]中的318.2 GPa和文献[37]中的323.2 GPa非常一致。根据经验知道,当某种材料的剪切模量与体弹模量的比值大于0.5时,该材料为脆性材料,且该比值越大脆性越大;若该比值小于0.5时,此材料为塑性材料,该比值越小延展性越好。从表3中可以看到,当钨含有缺陷时,除了钨中含有氢的替位掺杂和钨的自间隙原子这两种缺陷时向塑性转变,当钨中含有其他点缺陷时朝着脆性增加的方向转变。值得注意的是,当钨中含有氦的四面体点缺陷时,此时钨的剪切模量与体弹模量的比值最大为0.517,表明当钨中存在该种点缺陷时钨呈现为脆性。
表3钨及其含有缺陷时的体弹模量和剪切模量
Table 3Bulk modulus and shear modulus of perfect and defect tungsten
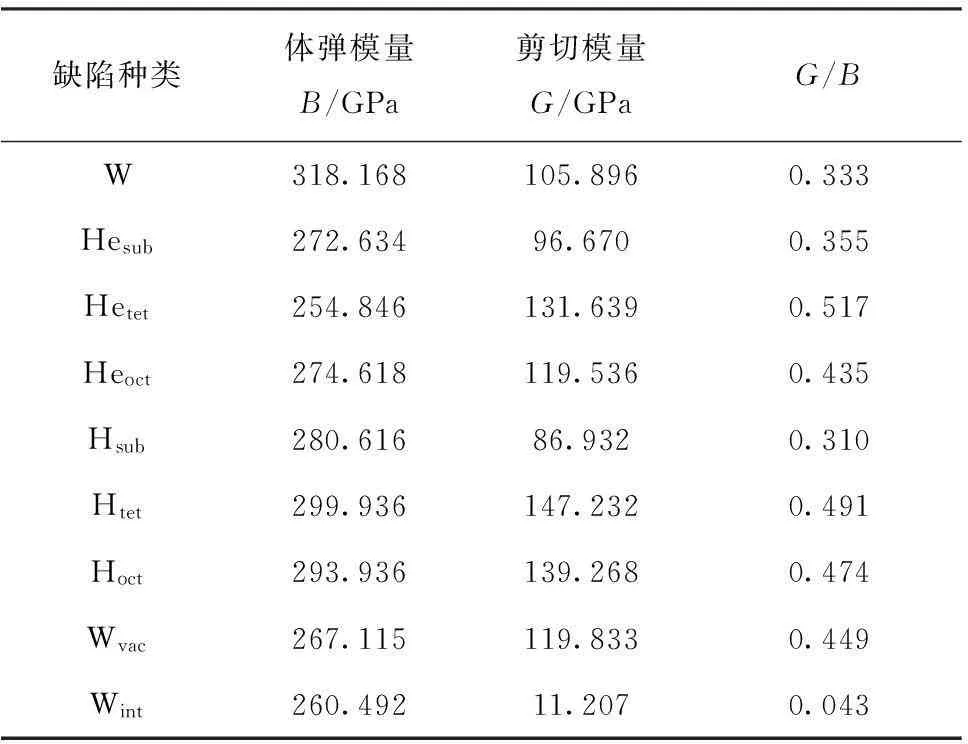
缺陷种类体弹模量B/GPa剪切模量G/GPaG/BW318.168105.8960.333Hesub272.63496.6700.355Hetet254.846131.6390.517Heoct274.618119.5360.435Hsub280.61686.9320.310Htet299.936147.2320.491Hoct293.936139.2680.474Wvac267.115119.8330.449Wint260.49211.2070.043
表4钨及其含有缺陷时的杨氏模量和泊松比
Table 4Young’s modulus and Poisson’s ratio of perfect and defect tungsten
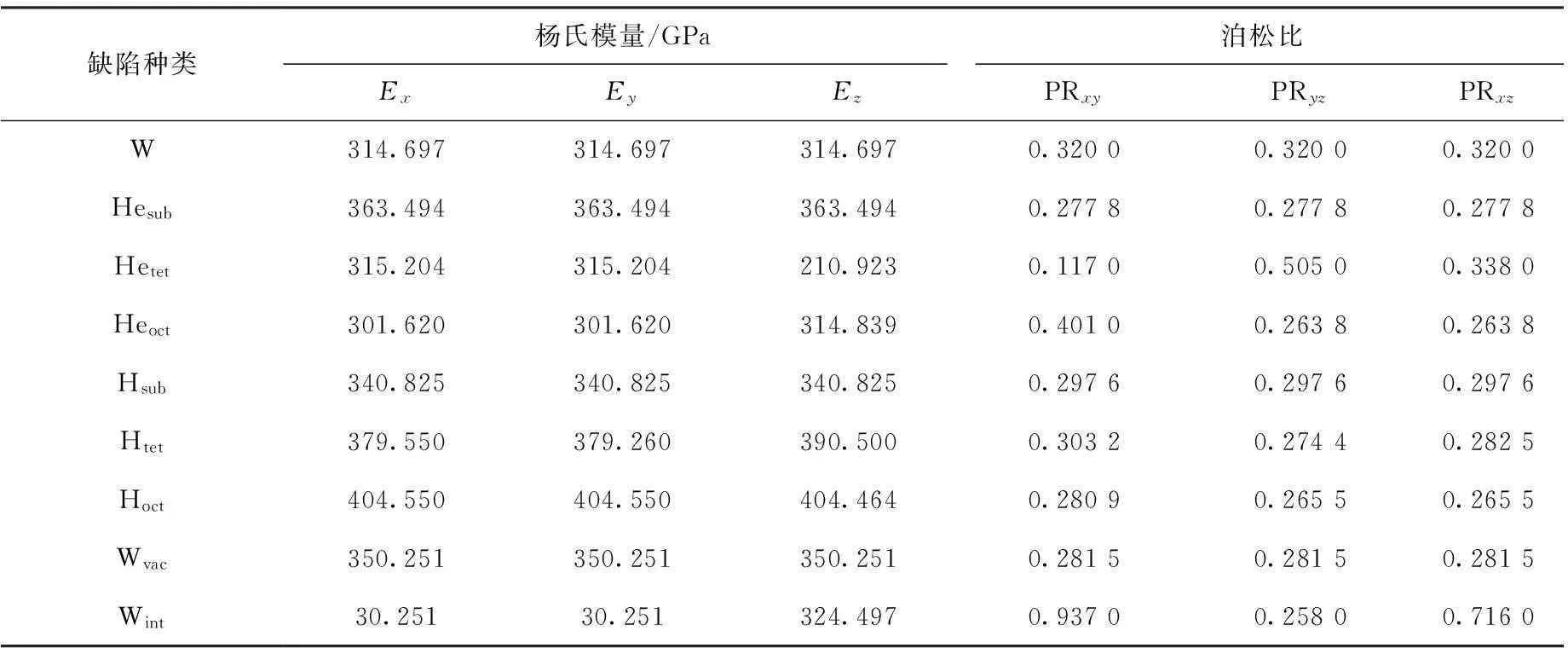
缺陷种类杨氏模量/GPa泊松比ExEyEzPRxyPRyzPRxzW314.697314.697314.6970.32000.32000.3200Hesub363.494363.494363.4940.27780.27780.2778Hetet315.204315.204210.9230.11700.50500.3380Heoct301.620301.620314.8390.40100.26380.2638Hsub340.825340.825340.8250.29760.29760.2976Htet379.550379.260390.5000.30320.27440.2825Hoct404.550404.550404.4640.28090.26550.2655Wvac350.251350.251350.2510.28150.28150.2815Wint30.25130.251324.4970.93700.25800.7160
在表4中可以看到,无论是杨氏模量还是泊松比,当钨中点缺陷在某些特定位置,材料表现出了各向异性。晶体发生向各向异性转变是氢或氦在钨中的四面体或八面体位置处形成的点缺陷所致,而氢、氦替位掺杂和空隙这三种点缺陷没有使钨在缺陷形成后向着各向异性转变。除了氢替位掺杂和自间隙原子这两种点缺陷外,其他带有点缺陷的钨的杨氏模量都有所增大,杨氏模量最大的为氢的八面体位掺杂。可以看出,在受到相同拉力时,氢氦存在于钨中时发生的形变量与纯钨相比会减小。从泊松比来看,含有点缺陷的钨的泊松比绝大多数向较小值转变。值得注意的是自间隙原子引起的各向异性也十分明显。无论是从杨氏模量还是泊松比的角度来看,氦的四面体位和八面体位掺杂引起的各向异性也较大。综上所述,当钨中含有点缺陷时,弹性常数的变化与缺陷在钨中的位置有相当大的关系,弹性常数的改变是因为缺陷晶体中原子间键的改变。
3结论
通过对含有氢或氦引起的点缺陷的钨进行第一性原理研究计算后发现:
(1) 氢氦在钨中形成点缺陷后,晶胞的体积有的增加有的减小,晶胞的体积变化与氢氦在钨中形成缺陷时所处位置有关。四面体和八面体处氢氦引起的点缺陷会引起晶体体积的增加,而氢氦替位掺杂钨会引起晶体体积的减小;
(2) 从形成能的角度来看,氦最容易形成替位掺杂,而氢最容易在四面体处掺杂形成点缺陷;
(3) 若钨中含有氢或氦引起的点缺陷,当缺陷为氢的替位或自间隙时晶体会向塑性改变,其余缺陷会使晶体向脆性方向转变。
(4) 缺陷的存在可能会使钨发生各向异性,当缺陷是替位或自间隙时不会发生各向异性,当氢或氦在四面体位时晶体的各向异性会较明显。钨中含有空位和自间隙原子这两种缺陷时,经比较发现自间隙这个缺陷引起的晶体变化十分明显,值得在今后的工作中注意。
本计算结果可为钨作为第一壁材料的研发工作提供理论参考。
参考文献:
[1]Wang X X, Zhang Y, Zhou H B, et al. Effects of niobium on helium behaviors in tungsten: a first-principles investigation[J]. Acta Physica Sinica, 2014, 63(4): 046103.
[2]Feng K M. Overview of the ITER test blanket module program[J]. Nucl Fusion Plasma Phys, 2006, 26(3): 161-169.
[3]Cho A. Bigger contribution to ITER erodes domestic fusion program[J]. Science, 2012, 335(6071): 901-902.
[4]丁文艺,何海燕,潘必才.磁约束可控热核聚变堆中的第一壁材料钨的研究状况和面临的若干问题[J].安徽师范大学学报(自然科学版),2014,37(4):314-319.
[5]Barabash V, Eaton R, Hirai T, et al. Summary of beryllium qualification activity for ITER first: wall applications[J]. Physica Scripta, 2011, T145: 014007.
[6]Conn R W, Doerner R P, Won J. Beryllium as the plasma-facing material in fusion energy systems-experiments, evaluation, and comparison with alternative materials[J]. Fusion Eng Des, 1997, 37: 481-513.
[7]Roth J, Eckstein W, Guseva M. Erosion of Be as plasma-facing material[J]. Fusion Eng Des, 1997, 37: 465-480.
[8]Patel B, Parsons W. Operational beryllium handling experience at JET[J]. Fusion Eng Des, 2003, 69: 689-694.
[9]Barabash V, Federici G, Lhnke N K J, et al. Material/plum surface interaction issues following neutron damage[J]. J Nucl Mater, 2003, 313-316: 42-51.
[10]Snead L, Jones R H, Kohyama A, et al. Status of silicon carbide composites for fusion[J]. J Nucl Mater, 1996, 233-237: 26-36.
[11]Fenici P, Rebelo A J F, Jones R H, et al. Current status of SiC/SiC composites R&D[J]. J Nucl Mater, 1998, 258-263: 215-225.
[12]Bolt H, Araki M, Linke J, et al. Heat flux experiments on first wall mock-ups coated by plasma sprayed B4C[J]. J Nucl Mater, 1996, 233-237: 809-813.
[13]Luo G N, Li Q, Zheng X B, et al. Coating materials for fusion application in China[J]. J Nucl Mater, 2011, 417: 1257-1261.
[14]Li H, Chen J L, Li J G, et al. High heat load tests on W/Cu mock-ups and evaluation of their application to EAST device[J]. Fusion Eng Des, 2009, 84: 1-4.
[15]Wang Y, Wang F C, Yu X D, et al. Effect of interlayer on stress wave propagation in CMC-RHA multi-layered structure[J]. Composites Science and Technology, 2010, 70: 1669-1673.
[16]刘彬彬,谢建新.W-Cu梯度热沉材料的成分与结构设计[J].稀有金属,2005,29(5):757-761.
[17]Gludovatz B, Wurster S, Hoffmann A, et al. Fracture toughness of poly crystalline tungsten alloys[J]. Int J Refract Met Hard Mater, 2010, 28: 674-678.
[18]种法力,陈俊凌,李建刚.VPS-EBW法制备W/Cu功能梯度材料及热负荷实验研[J].稀有金属材料工程,2006,35(9):1509-1512.
[19]Pintsuk G, Uytdenhouwen I. Thermo-mechanical and thermal shock characterization of potassium doped tungsten[J]. Int J Refract Met Hard Mater, 2010, 28: 661-668.
[20]Shu X Y, Qiu H X, Huang B, et al. Preparation and characterization of potassium doped tungsten[J]. J Nucl Mater, 2013, 440: 414-419.
[21]Zhang K, Shen W P, Ge C C. Effect of additives on thermal-shock resistance of W/Cu FGMs[J]. Multiscale and Functionally Graded Materials, 2008, 973: 838-843.
[22]Ritz G, Hirai T, Norajitra P, et al. Failure study of helium-cooled tungsten divertor plasma-facing units tested at DEMO relevant steady-state heat loads[J]. Physica Scripta, 2009, T138: 014064.
[23]Bazylev B, Landman I, Loarte A, et al. Experiments and modeling of droplet emission from tungsten under transient heat loads[J]. Physica Scripta, 2009, T138: 014061.
[24]Chiang W C, Baxter D V. Low-angle X-ray diffraction with insitu annealing: application to W/Cu multilayers[J]. J Appl Phys, 1993, 74(7): 4331-4338.
[25]Matejieek J, Boldyryeva H. Processing and temperature-dependent properties of plasma-sprayed tungsten-stainless steel composites[J]. Physica Scripta, 2009, 11138: 014041.
[26]Luo G N, Liu M, Kuang Z Q, et al. Directly-cooled VPS-W/Cu limiter and its preliminary results in HT-7[J]. J Nucl Mater, 2007, 363-365: 1241-1245.
[27]Li Q, Chen H, Qi P, et al. Modeling of heat deposition on the W/Cu movable limiter in HT-7[J]. Fusion Eng Des, 2010, 85: 126-129.
[28]Abhaszadeh H, Masoudi A, Safabinesh H, et al. Investigation on the characteristics of micro-and nano-structured W-15wt.% Cu composites prepared by powder metallurgy route[J]. Int J Refract Met Hard Mater, 2012, 30: 145-151.
[29]Fan J L, Liu T, Zhu S, et al. Synthesis of ultrafine/nanocrystalline W-(30-50) Cu composite powders and micro structure characteristics of the sintered alloys[J]. Int J Refract Met Hard Mater, 2012, 30: 33-37.
[30]Shimada T, Kikuchi H, Ueda Y, et al. Blister formation in tungsten by hydrogen and carbon mixed ion beam irradiation[J]. J Nucl Mater, 2003, 313-316: 204-208.
[31]Xu Jingcheng, Zhao Jijun. First-principles study of hydrogen in perfect tungsten crystal[J]. Nucl Instrum Method Phys Res, Sect B, 2009, 267: 3170-3174.
[32]Abdullah A, Liu Y L, Zhou H B, et al. First-principles investigation on dissolution and diffusion of oxygen in tungsten[J]. J Nucl Mater, 2009, 393: 508-512.
[33]Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple[J]. Phys Rev Lett, 1996, 77(18): 3865-3868.
[34]Tomaszewski P E. Structural phase transitions in crystals: I. Database[J]. Phase Transitions, 1992, 38(3): 127-220.
[35]Liu Y L, Zhang Y, Luo C N, et al. Structure, stability and diffusion of hydrogen in tungsten: a first-principles study[J]. J Nucl Mater, 2009, 390: 1032-1034.
[36]Landolt-Börnstein E P. Numerical data and functional relationship in science and technology[M]. New Series, Vol.Ⅲ. Heidelberg: Springer, 1991: 25.
[37]Kettle C. Introduction to solid states physics[M]. 6th Ed. New York: Wiley, 1986: 547.
收稿日期:2015-10-10;
修订日期:2015-11-28
基金项目:中央高校基本科研业务费专项资金资助项目(2014MS53)
作者简介:张峥(1990—),男,四川平昌人,硕士研究生,核能科学与工程专业 *通信联系人:赵强(1982—),男,北京人,博士,讲师,粒子物理与原子核物理专业,E-mail: qzhao@ncepu.edu.cn
中图分类号:TL341
文献标志码:A
文章编号:0253-9950(2016)01-0019-06
doi:10.7538/hhx.2016.38.01.0019
First-Principles Study on Effect of Hydrogen and Helium in Tungsten
ZHANG Zheng1, ZHAO Qiang1,*, OUYANG Xiao-ping1,2,3
1.Beijing Key Laboratory of Passive Safety Technology for Nuclear Energy, School of Nuclear Science and Engineering, North China Electric Power University, Beijing 102206, China;2.Northwest Institute of Nuclear Technology, Xi’an 710024, China;3.School of Materials Science and Engineering, Xiangtan University, Xiangtan 411105, China
Abstract:The formation energy of all kinds of point defects formed by hydrogen or helium and the change of elastic constants of tungsten containing a point defect were studied by using first-principles method based on GGA with a PBE form. The other two kinds of point defects, such as vacancy and self interstitial atom in tungsten, were studied by using the same method. After calculation, we found that the crystal volume has some changes due to the formation of a point defect in the tungsten formed by hydrogen or helium. And the results depend on the sites of point defects. The crystal volume becomes bigger when the defects are located in the tetrahedron or octahedron sites, while the volume becomes smaller when a tungsten atom is replaced by a hydrogen or helium atom. For helium, the substitution site is the most favorable over the tetrahedron and octahedron sites. For hydrogen, on the contrary, the tetrahedron site is the most perfect site over the substitution and octahedron sites. In these defects, we can find the smallest formation energy corresponds to the defect formed hydrogen which located in tetrahedron site, and the biggest formation energy to the formation of a self interstitial atom. If tungsten contains a point defect formed by hydrogen or helium, the body modulus and the shear modulus will have some changes. The crystal will change to plastic when there is a hydrogen substitute defect or self interstitial atom in tungsten. The tungsten becomes more brittle when it contains other point defects, but it still has the ductility in total. The crystals may become anisotropy when there is a defect in tungsten, and the results depend on the site of the defects. Only for the defects at the substitute site, the anisotropy will not happen. The research can provide a theoretical reference for the development of the plasma facing materials.
Key words:hydrogen; helium; tungsten; first-principles
