全基因组关联分析方法的研究进展
2016-07-11郝兴杰张淑君
郝兴杰,胡 林,张淑君
(华中农业大学动物科学技术学院/动物医学院 动物遗传育种与繁殖教育部重点实验室,武汉 430070)
全基因组关联分析方法的研究进展
郝兴杰,胡林,张淑君*
(华中农业大学动物科学技术学院/动物医学院 动物遗传育种与繁殖教育部重点实验室,武汉 430070)
摘要:全基因组关联分析目前已经成为研究复杂性状和疾病遗传变异的有效方法,但是由于群体结构的存在,导致分析结果出现虚假关联。经过数十年的发展,各种新方法不断出现和完善,用于减少群体结构对分析的影响。本综述将对在全基因组关联分析中能够处理群体结构的方法进行介绍,以期为进一步选择GWAS方法准确揭示各种性状的遗传背景提供参考。
关键词:全基因关联分析;群体结构;虚假关联
全基因组关联分析(Genome-wide association study,GWAS)是一种在全基因组范围内,通过高密度单核苷酸多态性(SNP)挖掘影响表型性状(如疾病,身高)基因的统计分析方法[1]。自从2005年,R.J.Klein等[2]利用GWAS第一次成功鉴定了影响年龄相关性视网膜黄斑变异的重要遗传因子之后,掀起了利用GWAS揭示复杂性状遗传基础的热潮,越来越多地被用来揭示人类以及动植物的常见疾病和复杂性状的遗传机理。目前GWAS主要采用两种试验设计,一种是基于无关个体的病例-对照(Case-control)设计[3-4],假设受试个体来源于单一群体,且个体之间互不相关,然而试验中无论如何控制,不同程度的亲缘关系和群体分层等群体结构都是无法避免的,尤其是在样本数量巨大的情况下;另一种是基于有亲缘关系的群体(Population-based cohorts)设计[5-6],假设受试个体来源于不同群体,在同一群体内的个体之间存在一定的亲缘关系,这种情况在动植物研究中极为常见。
在进行GWAS时,要求遗传背景一致或者相似的群体,然而无论采用哪种设计,试验个体都会面临着群体分层和亲缘关系等群体结构导致虚假关联(Spurious association)的结果。群体结构指来源于不同组别、群体或者地理区域的个体存在遗传差异,会导致群体之间的等位基因频率不同,表现为群体分层和亲缘关系。在进行关联分析时,如果忽略了群体结构的影响,将可能导致分析结果出现偏差,增加了假阳性错误的产生风险,在合并的群体中显著的SNP在各个群体中并不显著[7],如图1。
由于存在群体结构,不同亚群(Sub-population)之间基因交流的频率很低,等位基因频率存在差异性,在合并群体中,基因型的频率出现偏离哈代温伯格平衡检测(Hardy-Weinberg equilibrium test,HWE)预测值,这种现象称为华伦德效应(Wahlund effect)[8]。在对遗传疾病和复杂性状进行全基因组关联分析时,首先要进行的就是对SNP进行质控,其中就包括HWE,如果存在群体结构,尤其是不同亚群的遗传差异性较小,就需要增加样本数量来检测由于华伦德效应导致的HWE偏移。
在过去十几年内,出现了许多处理群体结构的GWAS方法,主要分为4种,分别为基因组控制法(Genomic control)、分层分析法(Stratification analysis)、主成分分析法(Principal components analysis,PCA)和混合线性模型分析法(Mixed-linear-model association,MLMA)。本文将对目前能够处理群体结构的GWAS方法作一综述,并对GWAS的方法研究进行展望,以期为进一步选择GWAS方法,准确揭示各种性状的遗传基础提供参考。
1基因组控制法
B.Devlin等最早提出利用基因组控制法来衡量群体结构对关联分析的影响[9-10]。在进行GWAS时,标准的关联检验方法一般为卡方检验或者趋势卡方检验(Armitage’s trend test),当群体结构存在时,统计量服从λχ2分布,其中λ为基因组膨胀因子(Genomic inflation factor),大小由群体结构控制,同时也受样本数量影响,可以用来衡量群体结构对关联分析的影响程度。在基因组控制法中,通过对原始的关联统计量统一除以λ得到新的统计量,实现对群体结构的校正。基因组膨胀因子λ可以通过基因组数据进行估测,假定选择了一组非关联的位点,那么每个位点的检验统计量服从λχ2分布,期望为λ,原假设为随机变量的统计量服从自由度为1的χ2分布,期望为1,由于中位数比平均数更加稳健,在实际计算过程中多采用中位数进行比较,膨胀因子的计算公式:
尽管基因组控制法能够处理群体结构造成的影响,但也有一些限制。对原始统计量统一进行校正,在一定程度上降低了检验的功效,尤其当群体结构的影响很大时,基因组控制法就比较保守[11-12]。根据经验,当λ<1.01时,认为群体结构影响很小;当1.01<λ<1.05时,认为群体结构的影响中等,但仍然在接受的范围之内;当λ>1.1时,表明群体结构影响很大,基因组控制法缺少检验的功效,需要选择其他方法对群体结构的影响进行校正[13]。
2分层分析法
在进行GWAS时样本可能混合了多个有遗传差异的亚群,对于亚群的划分可以根据地理区域、体型特征、经纬度等标准。然而这种划分方法比较主观,不能准确反映群体结构。如何准确的将试验个体聚类分群并与遗传信息相匹配,将有利于进一步研究相关问题。J.K.Pritchard等提出,在病例-对照设计中可以利用非关联的分子标记去检测群体结构[14]。随后,J.K.Pritchard等[15]采用贝叶斯聚类分析方法,假设有k个亚群,利用基因组上等位基因的基因型信息将受试个体分别指定到各个亚群中,并开发出相应的程序STRUCTURE。在对群体进行划分之后,可以在亚群之内进行关联分析,J.K.Pritchard等[16]认为,经过分群之后,亚群内将不再存在群体结构,关联分析将不再受群体结构的影响,不会出现虚假关联结果。
分层分析法的重要一步是对样本进行聚类分群,由于样本群体的遗传差异可能是连续的,分界不是绝对的,有些受试个体可能会被聚类到多个亚群中[15],该方法在用于全基因组大数据时,由于计算量太大限制了其使用[12]。D.H.Alexander等对STRUCTURE的贝叶斯方法中的最大似然估计进行优化,开发了ADMIXTURE程序,提高了计算效率,使其在聚类时可以适用于更多的标记[17]。
3主成分分析法
主成分分析在应用GWAS之前主要是作为一种数据降维的技巧,将大量相关变量转换成一组很少的不相关变量,这些无关变量被称为主成分,通过主成分尽可能多的解释初始变量的变异程度。N.Patterson等[18]根据主成分分析的思想,利用受试个体之间的亲缘关系来研究样本的群体结构。具体步骤包括:首先,根据全基因组等位基因频率构建亲缘关系矩阵(Kinship matrix);然后,计算亲缘关系矩阵的特征值和特征向量,特征向量揭示了样本中的非随机成分,即群体结构;最后,根据特征值的大小选择几个特征向量代替亲缘关系矩阵。A.L.Price等[12]将主成分分析应用于GWAS,首先通过主成分分析基因型数据特征值和特征向量,然后用特征向量对初始基因型和表型进行校正,最后采用卡方趋势检验对校正后的基因型和表型进行关联分析。
主成分分析和分层分析有很多联系和区别,分层分析的目的是通过聚类将样本分成明确的k个亚群,更直观的解释了群体结构,而主成分分析没有具体的模型,可适用于亚群分界不明显的样本群体,通过特征向量反映个体间的遗传差异是连续的,相较于分层分析更加稳健[17-18]。在进行主成分分析时,各个特征向量是正交的,A.L.Price等[12]发现,特征向量的使用数目对初始基因型和表型校正影响不显著,默认的特征向量使用数目为10个,使用过多的特征向量去校正反而会减低检验的功效,N.Patterson等[18]建议采用k个显著的特征向量去校正初始基因型和表型。
4混合线性模型法
上述提到的基因组控制法、分层分析法和主成分分析法主要是为了校正群体结构中的群体分层的影响,但是对于群体结构中的亲缘关系的影响上述3种方法都存在不足[19-20]。混合线性模型法[21]在常规遗传育种中用于最佳线性无偏预测(Best linear unbiased prediction,BLUP)动物的育种值,可以直接将两两个体之间的亲缘关系整合到模型中,考虑各种固定因素和随机因素的效应,J.Yu等[11]首次将混合线性模型用于GWAS,可以很好的控制群体结构的影响,降低虚假关联的产生并保持较高的检验功效。在GWAS中混合线性模型:
y=Xβ+Zu+e
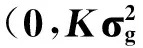
在混合线性模型中,最重要也是最耗时的一步就是方差组分的估计。J.Yu等研究的方法在对每个标记进行关联分析时,都需要对方差组分重新迭加估计,计算压力过大仅适用于样本量较少时进行GWAS,在样本量较大时利用混合线性模型不能够实现GWAS。W.M.Chen等[22]发现,当SNPs的效应很小时,方差组分可以近似等于全部SNPs估计的方差组分,只需要估计一次,使得混合线性模型法可以适用于大样本的GWAS。不断有研究者优化混合线性模型应用于GWAS的算法,采用两步计算的策略(Two-stage approach)将方差组分估计与关联分析分开,开发出新的GWAS方法,其中包括GRAMMAR[23]、EMMA[24]、GAPIT[25]、EMMAX[26]、GRAMMAR-Gamma[27]、Fast-LMM[28]、GCTA[29-30]和GEMMA[31]等方法,在这些方法中,GRAMMAR、GAPIT、EMMAX、GRAMMAR-Gamma属于近似算法,其他几种方法属于精确算法。
相较于基因组控制法、分层分析法和主成分分析法,混合线性模型法应用于GWAS有许多优势,但也存在一些不足和需要进一步改进的地方[19-20],例如在构建亲缘关系矩阵时如何选择全基因组上的分子标记才可以准确估计群体结构,在混合线性模型法运用于病例对照设计时会降低检验功效,将候选标记用于构建亲缘关系矩阵也将降低检验功效。M.Pirinen等[32]发现,将混合线性模型用于病例-对照设计时,在混合线性模型中添加一些已知的协变量作为固定效应时,如果是常见疾病(发病率高于20%),将会提高检验功效,但是对于罕见疾病,将会降低其检验功效,不能检测出新的遗传标记。在混合线性模型中,多基因效应和群体结构都会使GWAS的统计分布出现膨胀,现有的方法不能区别膨胀是来源于多基因效应还是群体结构造成的偏差,B.K.Bulik-Sullivan等[33]基于这点开发了新的方法“连锁不平衡评分回归”分析法(LD score regression),也证实了在大样本的GWAS中,统计分布的膨胀主要是由多基因效应造成的。现有的混合线性模型法大部分都建立在混合线性模型为无穷小的模型(Infinitesimal model)的假设之上,即所有的标记的影响都很小且都服从独立的正态分布,但实际上有影响的标记可能只有几千个,采用贝叶斯方法,可以区别效应大和效应小的标记,P.R.Loh等[34]基于这种情况,采用非无穷小的混合线性模型并优化了算法BOLT-LMM,减少了计算过程中的迭代次数,也提高了检验功效。
5展望
GWAS目前作为一种研究复杂性状和疾病遗传机理的重要方法,经过数十年的发展,各种方法不断出现和完善,混合线性模型法在处理群体结构上具有很大优势,目前被广泛运用于GWAS中。由于在动物群体中一般都存在较复杂的群体结构,我们应该结合群体结构和研究目的选择合适的GWAS方法。现有的GWAS方法大部分只考虑了加性效应,在进行关联分析时采用的单位点分析(Single-maker association)将导致一些稀有突变(Rare mutation)不能被有效检测出,在今后的GWAS方法研究中,多位点分析[35-36]、非加性效应以及互作效应[37]对GWAS的影响都应该成为一个重要的研究方向。
参考文献(References):
[1]RISCH N,MERIKANGAS K.The future of genetic studies of complex human diseases[J].Science,1996,273(5281):1516-1517.
[2]KLEIN R J,ZEISS C,CHEW E Y,et al.Complement factor H polymorphism in age-related macular degeneration[J].Science,2005,308(5720):385-389.
[3]CHARLIER C,COPPIETERS W,ROLLIN F,et al.Highly effective SNP-based association mapping and management of recessive defects in livestock[J].NatGenet,2008,40(4):449-454.
[4]SIRONEN A,UIMARI P,NAGY S,et al.Knobbed acrosome defect is associated with a region containing the genes STK17b and HECW2 on porcine chromosome 15[J].BMCGenomics,2010,11(699):1471-2164.
[5]PREISSLER R,TETENS J,REINERS K,et al.A genome-wide association study to detect genetic variation for postpartum dysgalactia syndrome in five commercial pig breeding lines[J].AnimGenet,2013,44(5):502-508.
[6]AI H,XIAO S,ZHANG Z,et al.Three novel quantitative trait loci for skin thickness in swine identified by linkage and genome-wide association studies[J].AnimGenet,2014,45(4):524-533.
[7]TEO Y Y.Common statistical issues in genome-wide association studies:a review on power,data quality control,genotype calling and population structure[J].CurrOpinLipidol,2008,19(2):133-143.
[8]WAHLUND S.Zusammensetzung von Populationen und Korrelationserscheinungen vom Standpunkt der Vererbungslehre aus betrachtet[J].Hereditas,1928,11(1):65-106.
[9]DEVLIN B,ROEDER K.Genomic control for association studies[J].Biometrics,1999,55(4):997-1004.
[10]DEVLIN B,ROEDER K,WASSERMAN L.Genomic control,a new approach to genetic-based association studies[J].TheorPopulBiol,2001,60(3):155-166.
[11]YU J,PRESSOIR G,BRIGGS W H,et al.A unified mixed-model method for association mapping that accounts for multiple levels of relatedness[J].NatGenet,2006,38(2):203-208.
[12]PRICE A L,PATTERSON N J,PLENGE R M,et al.Principal components analysis corrects for stratification in genome-wide association studies[J].NatGenet,2006,38(8):904-909.
[13]ZEGGINI E,MORRIS A.Analysis of complex disease association studies:a practical guide[M].Academic Press,2010.
[14]PRITCHARD J K,ROSENBERG N A.Use of unlinked genetic markers to detect population stratification in association studies[J].AmJHumGenet,1999,65(1):220-228.
[15]PRITCHARD J K,STEPHENS M,DONNELLY P.Inference of population structure using multilocus genotype data[J].Genetics,2000,155(2):945-959.
[16]PRITCHARD J K,STEPHENS M,ROSENBERG N A,et al.Association mapping in structured populations[J].AmJHumGenet,2000,67(1):170-181.
[17]ALEXANDER D H,NOVEMBRE J,LANGE K.Fast model-based estimation of ancestry in unrelated individuals[J].GenomeRes,2009,19(9):1655-1664.
[18]PATTERSON N,PRICE A L,REICH D.Population structure and eigenanalysis[J].PLoSGenet,2006,2(12):e190.
[19]PRICE A L,ZAITLEN N A,REICH D,et al.New approaches to population stratification in genome-wide association studies[J].NatRevGenet,2010,11(7):459-463.
[20]YANG J,ZAITLEN N A,GODDARD M E,et al.Advantages and pitfalls in the application of mixed-model association methods[J].NatGenet,2014,46(2):100-106.[21]HENDERSON C.Application of linear models in animal breeding[D].Guelph:University of Guelph,1984.
[22]CHEN W M,ABECASIS G R.Family-based association tests for genomewide association scans[J].AmJHumGenet,2007,81(5):913-926.
[23]AULCHENKO Y S,DE KONING D J,HALEY C.Genomewide rapid association using mixed model and regression:a fast and simple method for genomewide pedigree-based quantitative trait loci association analysis[J].Genetics,2007,177(1):577-585.
[24]KANG H M,ZAITLEN N A,WADE C M,et al.Efficient control of population structure in model organism association mapping[J].Genetics,2008,178(3):1709-1723.
[25]ZHANG Z,ERSOZ E,LAI C Q,et al.Mixed linear model approach adapted for genome-wide association studies[J].NatGenet,2010,42(4):355-360.
[26]KANG H M,SUL J H,SERVICE S K,et al.Variance component model to account for sample structure in genome-wide association studies[J].NatGenet,2010,42(4):348-354.
[27]SVISHCHEVA G R,AXENOVICH T I,BELONOGOVA N M,et al.Rapid variance components-based method for whole-genome association analysis[J].NatGenet,2012,44(10):1166-1170.
[28]LIPPERT C,LISTGARTEN J,LIU Y,et al.FaST linear mixed models for genome-wide association studies[J].NatMethods,2011,8(10):833-835.
[29]YANG J,MANOLIO T A,PASQUALE L R,et al.Genome partitioning of genetic variation for complex traits using common SNPs[J].NatGenet,2011,43(6):519-525.
[30]YANG J,LEE S H,GODDARD M E,et al.GCTA:a tool for genome-wide complex trait analysis[J].AmJHumGenet,2011,88(1):76-82.
[31]ZHOU X,STEPHENS M.Genome-wide efficient mixed-model analysis for association studies[J].NatGenet,2012,44(7):821-824.
[32]PIRINEN M,DONNELLY P,SPENCER C C.Including known covariates can reduce power to detect genetic effects in case-control studies[J].NatGenet,2012,44(8):848-851.
[33]BULIK-SULLIVAN B K,LOH P R,FINUCANE H K,et al.LD Score regression distinguishes confounding from polygenicity in genome-wide association studies[J].NatGenet,2015,47(3):291-295.
[34]LOH P R,TUCKER G,BULIK-SULLIVAN B K,et al.Efficient Bayesian mixed-model analysis increases association power in large cohorts[J].NatGenet,2015,47(3):284-290.
[35]KORTE A,VILHJALMSSON B J,SEGURA V,et al.A mixed-model approach for genome-wide association studies of correlated traits in structured populations[J].NatGenet,2012,44(9):1066-1071.
[36]SEGURA V,VILHJALMSSON B J,PLATT A,et al.An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations[J].NatGenet,2012,44(7):825-830.
[37]THOMAS D.Gene-environment-wide association studies:emerging approaches[J].NatRevGenet,2010,11(4):259-272.
(编辑郭云雁)
Progresses in Research of Genome-wide Association Study Methods
HAO Xing-jie,HU Lin,ZHANG Shu-jun*
(KeyLaboratoryofAnimalBreedingandReproductionofMinistryofEducation,CollegeofAnimalScienceandTechnology/CollegeofVeterinaryMedicine,HuazhongAgriculturalUniversity,Wuhan430070,China)
Key words:genome-wide association study (GWAS);population structure;spurious association
Abstract:The genome-wide association study (GWAS) has become an effective approach to identify genetic variants associated with complex traits and diseases.However,population structure can result in spurious association.In the past few decades,new approaches were developed and improved to minimize the influence of population structure.In this review,we summarize some new approaches to treat population structure for selecting the best method for any GWAS to reveal the genetic backgroud of some traits.
doi:10.11843/j.issn.0366-6964.2016.02.001
收稿日期:2015-06-01
基金项目:促进与美大地区科研合作与高层次人才培养项目(52902-0650104);欧盟FPT构架项目玛丽居里夫人人才基金(Marie Curie Action,P11FR-GA-2012-912205)
作者简介:郝兴杰(1990-),男,湖北南漳人,博士,主要从事动物遗传疾病的研究,E-mail:xingjiehao@webmail.hzau.edu.cn *通信作者:张淑君,教授,E-mail:sjxiaozhang@mail.hzau.edu.cn
中图分类号:S813.3
文献标志码:A
文章编号:0366-6964(2016)02-0213-05
