铂催化吲哚丙炔酯合成咔唑类化合物*
2016-06-05张宁王德伟
石 赟,张宁,王德伟
(兰州大学 化学化工学院,甘肃 兰州 730000)
芳环和杂环化合物在有机合成和药物合成中占有主导性地位。因而如何高效地设计和合成此类化合物始终是有机化学工作者所面临的挑战之一。咔唑类化合物作为重要的含氮杂环化合物,是煤焦油中经济价值最高的成分之一。此结构在具有生物活性的杂环化合物合成中具有重要的作用。2009年,Benjamin J Stokes小组以二联芳基叠氮为原料,在Rh2(Ⅱ)的催化作用下合成咔唑类化合物[1]。2014年,Kazutaka Takamatsu小组又以二氨基联苯为原料,通过Cu(OAc)2催化分子内C—H/N—H偶联来合成咔唑[2]。2015年,Michael J James小组以3-炔醇吲哚为原料,以Ag(Ⅰ)为催化剂成功合成了咔唑类化合物[3]。
作者以N-甲基-3-吲哚丙炔酯为反应底物,讨论这类反应的可能性。考察了不同过渡金属催化剂对反应的影响,并探讨了反应溶剂、温度、反应时间对该反应分离产率的影响。
1 实验部分
1.1 试剂与仪器
氢氧化钾、二甲基亚砜(DMSO)、N,N-二甲基甲酰胺、无水乙醚、四氢呋喃、甲苯、苯甲醚、1,4-二氧六环、乙腈、1,2-二氯乙烷、二氯甲烷:分析纯,利安隆博华(天津)医药化学有限公司;吡啶:分析纯,上海中泰化学试剂有限公司。
实验中未经说明的药品和溶剂均为市售分析纯试剂,所有反应均用薄层硅胶板(GF254)进行TLC监测跟踪,产物利用硅胶柱纯化(200~300 μm),部分硅胶柱需要用三乙胺压洗,所用洗脱剂为工业乙酸乙酯与工业石油醚(沸程60~90 ℃)的混合液。
1H NMR和13C NMR使用AM-400核磁共振波谱仪:溶剂选用CDCl3,内标为TMS,德国Bruker公司;质谱使用[70eV(EI)]质谱仪:ZAB-HS,英国VG公司。
1.2 吲哚丙炔酯的制备
1.2.1 氮保护基吲哚的制备
在50 mL的圆底烧瓶中加入相应的吲哚(5.0 mmol,0.586 g)和KOH(10.0 mmol,0.560 g),然后加入20 mL的DMSO,搅拌,再加入碘甲烷(10.0 mmol,1.420 g),室温下反应。TLC监测反应结束后,用20 mL水淬灭反应,用乙酸乙酯萃取3次,合并有机相,无水硫酸钠干燥,减压除去溶剂,粗产品柱色谱分离,得0.69 g黄褐色固体,产率100%[4],反应方程式如下。

1.2.2 N-甲基-3-吲哚甲醛的制备
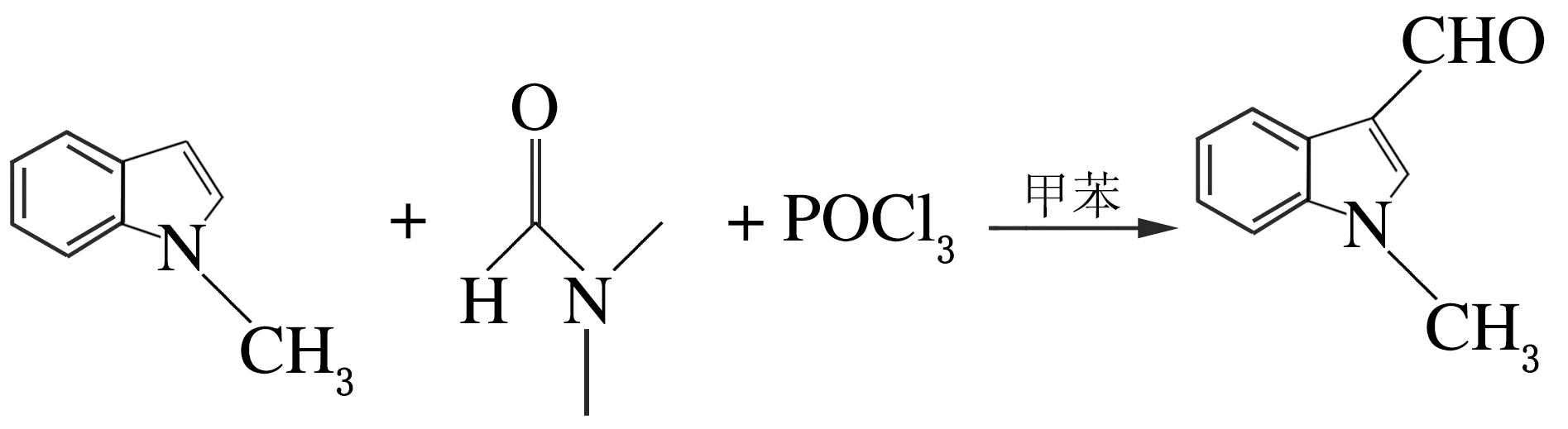
在100 mL的具支烧瓶中加入无水甲苯(10 mL),在氩气条件下滴加N,N-二甲基甲酰胺(4 mmol,0.330 g)。冷却至0~-5 ℃,缓慢滴加三氯氧磷(0.5 mL),滴毕,在室温下搅拌30 min,将N-甲基吲哚(5 mmol)溶于4 mL无水甲苯中,在惰气条件下缓慢滴加。然后在室温下反应3 h,加入4 mol/L NaOH溶液60 mL,加热回流,TLC检测,冷至室温,乙酸乙酯萃取3次,合并有机相,无水硫酸钠干燥,减压蒸馏,粗产品柱色谱分离,得0.443 2 g的黄褐色固体,产率59%[5],反应方程式如下。
1.2.3 N-甲基-3-吲哚丙炔醇的制备
氩气保护下,在50 mL的具支烧瓶中加入镁粉(5 mmol,0.120 g)和二氯化汞(0.05 mmol,0.014 g),加入无水乙醚做溶剂(5 mL),搅拌,缓慢滴加炔丙基溴(5 mmol,0.595 g)溶于无水乙醚(3 mL)的溶液,室温下反应1 h,直至反应液由黄色变为灰色。在冰盐浴条件下,把N-甲基-3-吲哚甲醛(2.5 mmol,0.403 g)溶于无水四氢呋喃(3 mL),在氩气下缓慢加入烧瓶中,反应1 h。反应结束后,用饱和氯化铵溶液将反应淬灭,乙酸乙酯萃取3次,合并有机相,硫酸镁干燥,减压蒸馏除去溶剂,快速柱色谱分离,即可得3-吲哚炔醇,黄色液体,产率62%[6],反应方程式如下。

1.2.4 N-甲基-3-吲哚丙炔酯的制备
在冰浴下,N-甲基-3-吲哚丙炔醇(1.5 mmol,0.302 g),吡啶(0.4 mL)和4-二甲氨基吡啶(0.33 mmol,40mg)加入50 mL圆底烧瓶中,缓慢加入醋酸酐(0.32 mL),反应6 h,加入饱和碳酸氢钠溶液(20 mL),二氯甲烷萃取3次,合并有机相,硫酸镁干燥,减压蒸馏除去溶剂,柱色谱分离即可得3-吲哚丙炔酯,产率91%[6],反应方程式如下。

1.3 N-甲基咔唑的制备合成
在10 mL的反应管中,依次加入称量好的N-甲基-3-吲哚丙炔酯(0.072 3 g,0.3 mmol),催化剂PtCl2[n(PtCl2)∶n(N-甲基-3-吲哚丙炔酯)=10%],然后加入甲苯溶液(4 mL),置于120 ℃的油浴中,维持此温度,搅拌反应4 h,TLC监测反应的进程。当反应完全后,停止加热,冷却至室温,用水淬灭,倒入分液漏斗中,用乙酸乙酯溶液萃取3次,然后合并有机相,用饱和的NaCl溶液洗涤反萃有机相,用硫酸镁干燥,减压蒸馏除去溶剂,柱色谱分离即可得到N-甲基咔唑。
2 结果与讨论
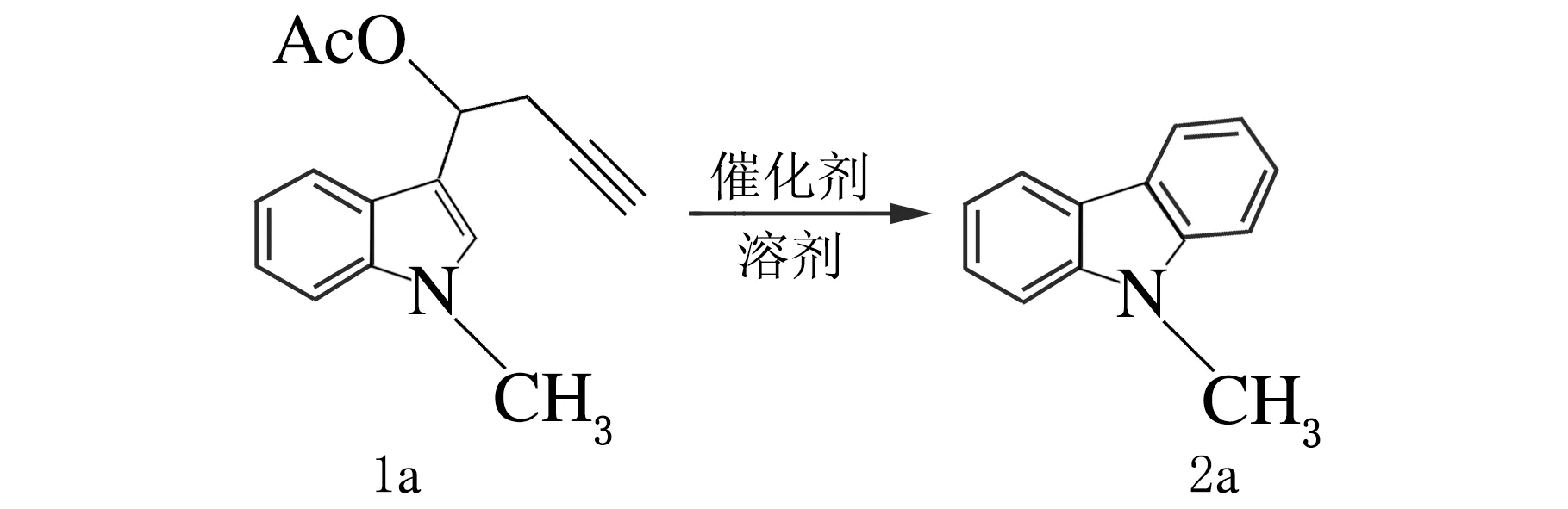
在实验的初始阶段,构建合成了化合物N-甲基-3-吲哚丙炔酯(1a),以其为底物讨论Pt催化吲哚丙炔酯反应的可能性,并对反应条件进行了筛选,反应方程式如下。
2.1 催化剂种类和溶剂对分离产率的影响
实验以1a为例进行了催化剂和溶剂的选择。考虑到空白实验的重要性,先进行了空白实验操作,在不添加任何催化剂的条件进行了实验,然后又选用了Pt(MeCN)2Cl2、 PtCl2、PtBr2、PtCl4和PtI4等不同的Pt催化剂[n(Pt催化剂)∶n(1 a) =10%]来进行尝试。对于大多数有机反应来说,溶剂的选择也非常重要,所以实验分别选用了甲苯、苯甲醚、1,2-二氯乙烷(DCE)、乙腈(MeCN)、1,4-二氧六环、二氯甲烷(DCM)等不同溶剂进行反应,以寻找具有最佳效果的溶剂,结果见表1。
研究发现在不添加催化剂的条件下没有产物生成,但加入Pt的催化剂均有产物生成,说明Pt催化剂都能催化这类环化反应的发生,其中PtCl2的催化效率最高,产率可达到67%。选择溶剂的实验表明,各种不同溶剂甲苯、苯甲醚、DCE、MeCN、1,4-二氧六环、DCM等对反应都有影响。而且,在PtCl2的催化下,这几种溶剂也都能使底物发生成环反应,用甲苯作溶剂时反应进行得最好。

表1 催化剂种类以及溶剂对分离产率的影响1)
1) 反应条件:1a(0.3 mmol),n(Pt催化剂)∶n(1a) =10%,溶剂4 mL,温度120 ℃,反应时间4 h。
2.2 温度与时间对分离产率的影响
随着所用催化剂、溶剂的确定,需要对反应温度与时间进行优化,所以进行了以PtCl2为催化剂、甲苯为溶剂,采用不同的温度与时间的实验,以寻找具有最佳效果的反应温度与时间,实验结果见表2。

表2 反应温度与时间对分离产率的影响
从表2可以看出,反应的最佳温度为120 ℃,低于120 ℃,产物的收率明显降低。而最适宜的反应时间为4 h,反应时间减短,产率有所降低,反应时间增长,产率并没有增加。
2.3 底物的拓展
经过对催化剂、溶剂、反应温度和时间的筛选,确定了n(PtCl2)∶n(1a) =10%,甲苯作溶剂,120 ℃条件下反应4 h,所生成的环化产物的产率最高可达67%,反应方程式如下。在得出最优反应条件之后,对该环化反应的底物适用范围进行了探讨,结果见表3。
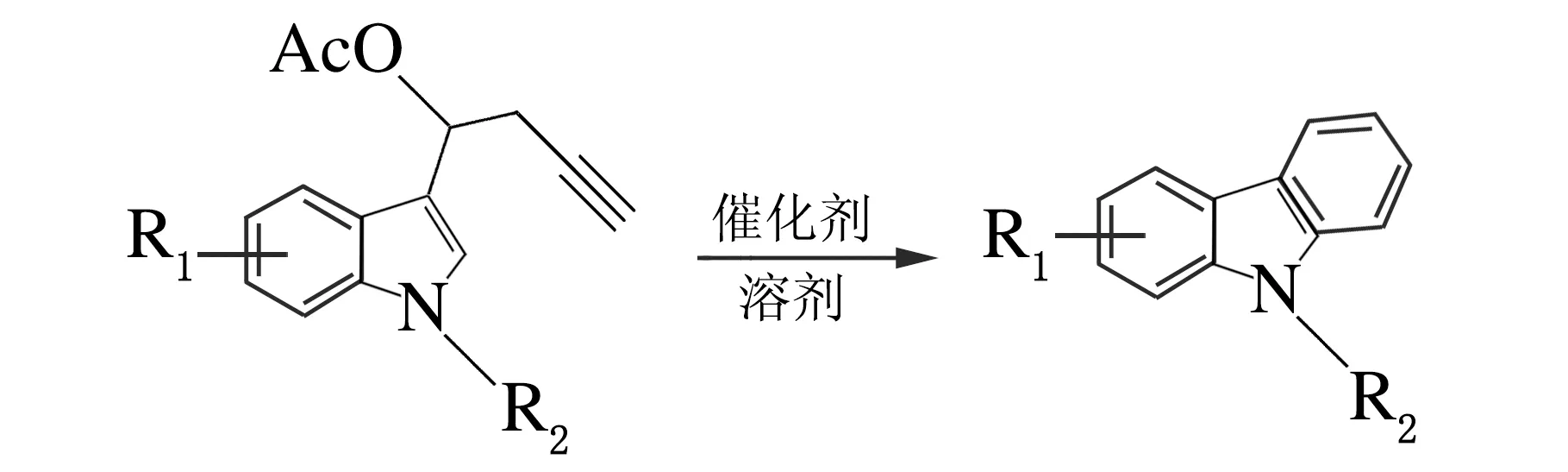
表3 底物的拓展1)

序号底物Ⅰ产物 Ⅱ产率/%1671a2a2<51b[5-7]2b3801c[5-6,8]2c4621d[4-6]2d5721e[5-6,8]2e6641f[4-6]2f7461g[4-6]2g
1) 反应条件:底物Ⅰ0.3 mmol,n(Pt催化剂)∶n(底物Ⅰ) =10%,溶剂4 mL,反应温度120 ℃,反应时间4 h。
表3合成了7个吲哚丙炔酯类化合物来拓展底物,发现吲哚上不同的取代基,吲哚N上不同的保护基团,都会对反应的产率产生一定的影响。吲哚上取代基的影响相对较小,而吲哚N上不同保护基团对反应影响较大,且发现保护基团的吸电子性越强,成环反应越容易进行、产率越高。
2.4 反应机理的推测
在查阅了一定的文献资料[9-10],并对其进行分析研究后,结合实验研究结果,通过相关的对照实验,提出了PtCl2参与的可能反应机理。首先是乙酰基的羰基进攻经金属铂活化过的炔基,形成反应中间体Ⅲ,接着羰基氧上的电子向炔基转移,形成环状过渡态金属卡宾中间体Ⅳ,随后炔基烯丙位的C—O断裂,电子重新排布,进一步与吲哚2位环化形成六元环,最后脱除一分子乙酸得到目标产物,反应方程式如下。
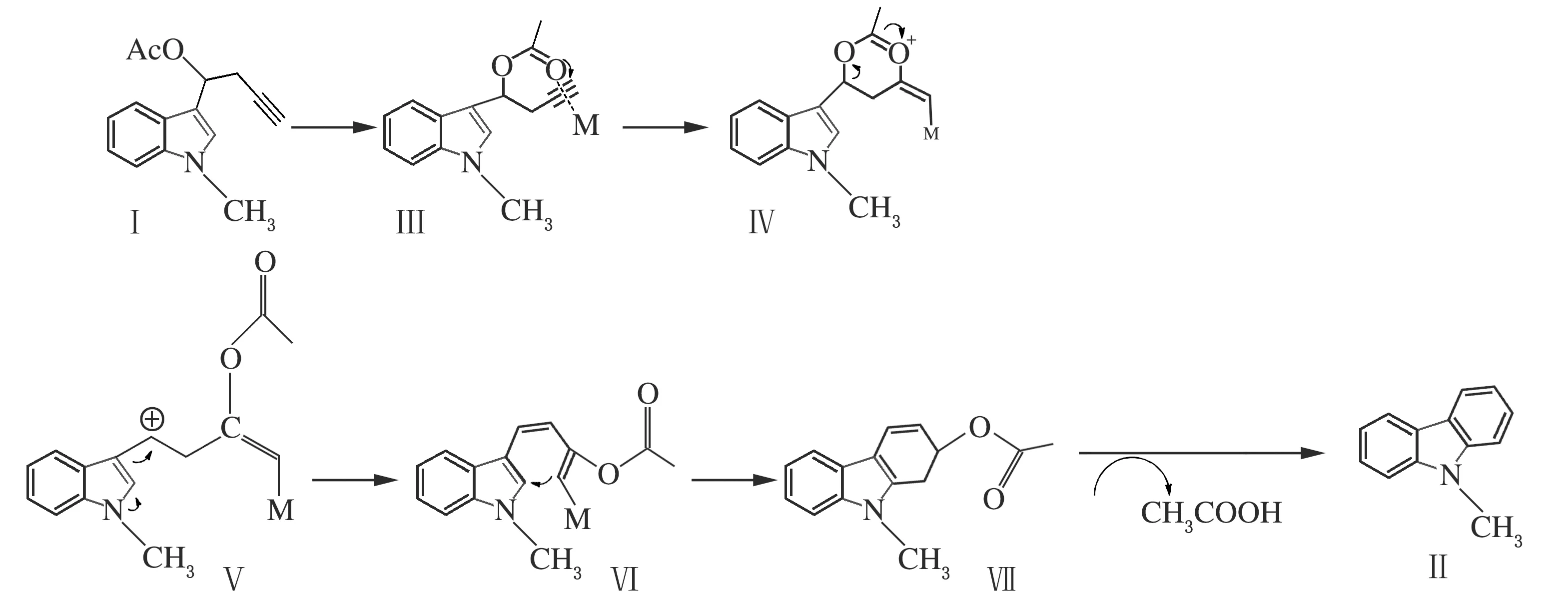
图1 可能的反应机理
3 结 论
实现了用吲哚丙炔酯在PtCl2催化条件下分子内成环生成咔唑的反应,以氯化铂为催化剂,甲苯做溶剂,120 ℃下反应4 h,可以达到67%的产率。反应过程简单,后处理方便,且底物适用性较好,是一种新的合成咔唑衍生物的实验方法,为其在药物、材料等领域的应用提供了新的合成思路。
参 考 文 献:
[1] BENJAMIN J STOKES,BRANKICA JOVANOVIC,DONG HUIJUN,et al.Rh2(Ⅱ)-catalyzed synthesis of carbazoles from biaryl azides[J].J Org Chem,2009,74 (8):3225-3228.
[2] KAZUTAKA TAKAMATSU,KOJI HIRANO,et al.Synthesis of carbazoles by copper-catalyzed intramolecular C—H/N—H coupling[J].Org Lett,2014,16 (11):2892-2895.
[3] MICHAEL J JAMES,ROSA E CLUBLEY,KLEOPAS Y PALATE,et al.Silver(Ⅰ)-catalyzed dearomatization of alkyne-tethered indoles:divergent synthesis of spirocyclic indolenines and carbazoles[J].Org Lett,2015,17(17):4372-4375.
[4] ZHANG L,PENG C,ZHAO D,et al.Cu(Ⅱ)-catalyzed C—H (SP3) oxidation and C—N cleavage:base-switched methylenation and formylation using tetramethylethylenediamine as a carbon source[J].Chem Commun,2012,48:5928-5930.
[5] BAGHER AMIR-HEIDARI,JENNY THIRLWAY,JASON MICKLEFIELD.Stereochemical course of tryptophan dehydrogenation during biosynthesis of the calcium-dependent lipopeptide antibiotics[J].Org Lett,2007,9(8):1513-1516.
[6] XU M,REN T T,LI C Y.Gold-catalyzed oxidative rearrangement of homopropargylic ether via oxonium ylide[J].Org Lett,2012,14(18):4902-4905.
[7] KEIGO KAMATA,JUN KASAI,KAZUYA YAMAGUCH,et al.Efficient heterogeneous oxidation of alkylarenes with molecular oxygen[J].Org Lett,2004,6(20):3577-3580.
[8] YU YOSHII,TAKANORI OTSU,NORIHIKO HOSOKAWA,et al.Synthetic studies toward penitrem E:enantiocontrolled construction of B-E rings[J].Chem Commun,2015,51:1070-1073.
[9] SANDRO CACCHI,GIANCARLO FABRIZI,et al.Palladium-catalyzed synthesis of 2-(aminomethyl) indoles from 3-(o-trifluoroacetamidoaryl)-1-propargylic alcohols and amines[J].J Org Chem,2014,79(1):401-407.
[10] ILARIA AMBROGIO,SANDRO CACCHI,GIANCARLO FABRIZI.Palladium-catalyzed synthesis of 2-(aminomethyl)indoles from ethyl 3-(o-trifluoroacetamidophenyl)-1-propargyl carbonate[J].Org Lett,2006,8(10):2083-2086.
Abstract: Carbazole compounds were synthesized by indole propargylic esters in the presence of PtCl2in toluene at 120 ℃ for 4 h.The chemical structures of these obtained compounds were characterized by MS,1H NMR and13C NMR.Based on the study of the reaction mechanism and influencing factors,we found that the N of indole bearing stronger electron-withdrawing groups participated well in the cyclization reaction.
Keywords: Indole propargylic esters;Carbazole;Transition metal catalysis;Cyclization reaction
