2-吲哚乙酸乙酯的合成工艺研究
2015-12-24陈芬,覃宇,李建芬等
通信作者:李建芬(1968-),男,博士,教授,E-mail:lijfen@163.com.
2-吲哚乙酸乙酯的合成工艺研究
陈芬1,覃宇1,李建芬2,王强胜2,熊志翀2
(1.武汉职业技术学院 生物工程学院,湖北 武汉 430074; 2.武汉轻工大学 化学与环境工程学院,湖北 武汉 430023)
摘要:探究2-吲哚乙酸乙酯新的合成方法和合成工艺。以2-硝基苯乙酸和丙二酸二乙酯为原料,通过亲核加成和还原环化反应,合成2-吲哚乙酸乙酯,关键合成步骤以不同的物料比、温度和反应时间进行反应,测得最终反应的转化率和产率,得到新的合成路线和最佳合成工艺。合成的最终产物为浅棕色固体,熔点为28—31 ℃,利用IR、1HNMR和MS等分析手段对中间体和终产物的结构进行了表征和分析,确认最终产物即为目标产物2-吲哚乙酸乙酯。新的合成方法反应条件温和,中间体产物无须分离,关键步骤最佳合成工艺为:物料配比n(2-硝基苯乙酸):n(丙二酸单乙酯钾盐)为1∶2,反应温度5 ℃,反应时间为20 h,此时产品收率达87.5%,色谱检测纯度≥98%。
关键词:吲哚;医药中间体;合成;表征;工艺
收稿日期:2015-03-26.修回日期:2015-05-11.
作者简介:陈芬(1966-),女,副教授,E-mail:chen25fen@163.com.
基金项目:湖北省教育厅科研项目(B20129202);武汉市青桐计划项目(2014042407111156).
文章编号:2095-7386(2015)03-0052-05
DOI:10.3969/j.issn.2095-7386.2015.03.011
中图分类号:O 626 32
Research on thesynthesis of 2-indoleacetic acid ethyl ester
CHENFen1,QINYu1,LIJian-fen2,WANGSheng-qiang2,XIONGZhi-chong2
(1.Biology Engineering Department,Wuhan Polytechnic,Wuhan 430074,China;
2. School of Chemical and Environmental Engineering, Wuhan Polytechnic University, Wuhan 430023, China)
Abstract:To explore the new method for synthesizing 2- indole acetic acid ethyl ester, and research the synthesis process. 2-indoleacetic acid ethyl ester is synthesized by the nucleophilic addition and the reductive cyclisation reactions using 2-nitrophenyl acetic acid and diethyl malonate as raw materials,a new synthetic route was obtain and the best synthesis processin were optimized, through the determination of the last reaction conversion rate and yield, with different raw material ratio, reaction temperature and reaction time reaction time and temperature of reaction in the key step of synthesis.The final product synthesis is light brown solid, the melting point of about 28-31℃, by using IR and 1HNMR, MS and other analytical methods, the structure of the intermediates and the end products were characterized and analyzed, confirming the final product is the target product 2-indoleacetic acid ethyl ester.The new synthesis method has the advantages of mild reaction conditions, intermediate products do not need to be separated,and the synthesis process of the key steps is best material ratio n (2- nitrophenyl acetic acid):n (ethyl malonate potassium salt) was 1:2, reaction temperature was 5 C, reaction time was 20h, at this condition ,the yield was 87.5%, the chromatographic purity was more than 98%.
Key words:indole; pharmaceutical intermediates; synthesis; characterization; research on process
1引言
吲哚类化合物约占生物碱的四分之一。近年来研究表明:2-吲哚乙酸乙酯是一种重要的医药中间体,由2-吲哚乙酸乙酯衍生的吲哚类化合物具有多种生理作用,如抗疟疾、抗肿瘤、抗糖尿病等,可用作5-羟色胺(HT)受体抑制剂、环氧酶抑制剂[1]。
吲哚类化合物的合成方法很多,一般是先引入所需的取代基,然后直接合成吲哚环。主要包括Fischer酸催化下σ移位重排环化反应、Pd-Cu催化的Sonogahira反应、Pd-zeohte催化反应、自由基环化合成、钌催化的苯胺与环氧烷开环合成吲哚等[2-9]。但是由于这些反应的条件较苛刻(如需要高温、强碱等),原料昂贵,反应步骤多,分离困难,且不具有广泛的官能团相容性,从而限制了它们在工业和医药等方面的应用。
迄今为止,尚未见由2-硝基苯乙酸和丙二酸二乙酯为原料,通过亲核取代和还原—缩合协同反合成2-吲哚乙酸乙酯及其衍生物的研究报道。
2材料与方法
2.1仪器与试剂
熔点测定仪(RY-1),红外分光光度计(WGH-30/30A),高效液相色谱仪(岛津LC-20AT),Bruker Avance 400 MHz核磁共振仪(内标为TMS,溶剂为DMSO、CDCl3)。
丙二酸二乙酯,邻硝基苯乙酸,N,N'-羰基二咪唑(CDI)(以上原料纯度均为98%以上)。
氢氧化钾,无水乙醇,三乙胺,乙腈,二甲亚砜,三氯甲烷,丙酮,乙醚,无水氯化镁,乙酸乙酯,碳酸氢钠,氯化钠,无水硫酸钠,醋酸铵,三氯化钛溶液(以上试剂均为分析纯)。
2.2实验方法
2.2.1合成方法
笔者采用一锅法合成2-吲哚乙酸乙酯,合成路线如图1所示。
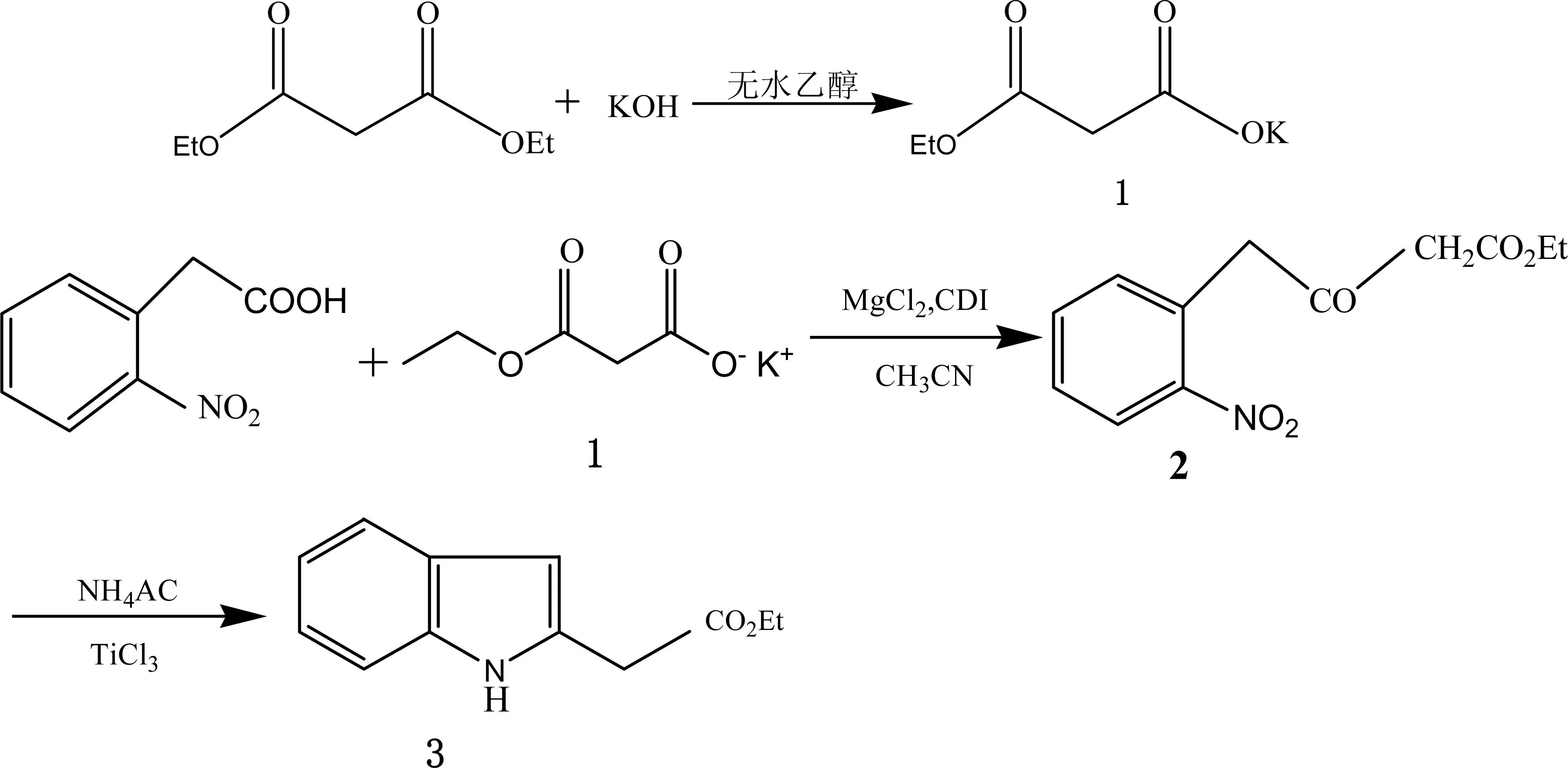
图1 2-吲哚乙酸乙酯的合成路线
2.2.2丙二酸单乙酯钾盐(化合物1)的制备[7]
在250 mL圆底三颈瓶中,加入无水乙醇100 mL,搅拌下缓慢加入氢氧化钾14.0 g(0.25 mol),温度不超过40 ℃,开启强力电动搅拌器,调整转速为150 r/min,搅拌冷却至室温,形成醇钾悬浊液。
将丙二酸二乙酯(40.0 g, 0.25 mol)加入到三口烧瓶中,再加50 mL无水乙醇,将醇钾悬浊液以2 mL/min滴加到三口烧瓶中,滴加完毕后,反应混合物继续在室温下搅拌3 h,得到白色沉淀物,过滤,收集并用冰的无水乙醇洗涤,真空干燥,得到白色固体41.7 g(产率98%)。无需纯化,进入下步反应。
2.2.32-硝基苯乙酰乙酸乙酯(化合物2)的
制备[8-9]
溶液a:将化合物1(14.00 g, 0.082 mol)和无水氯化镁9.32 g(0.979 mol)加入到100 mL无水乙腈中,搅拌制成悬浊液,再将三乙胺17.48 mL(0.125 mol)缓慢加入到此悬浊液中,混合物在室温下搅拌2 h,得溶液a。
溶液b:将邻硝基苯乙酸(7.05 g, 0.039 mol)和N, N'-羰基二咪唑(CDI) 6.97 g(0.043 mol)加入到40 mL无水乙腈中,搅拌15 min,形成溶液b。
将溶液b逐滴加入到溶液a中,反应混合物在0—5 ℃下搅拌18 h,然后升温至25 ℃回流2 h。反应结束后,在低于25 ℃下滴加100 mL的盐酸溶液(1.0 mol/L),搅拌15 min,分离出有机层,减压浓缩。浓缩物用100 mL乙酸乙酯溶解。水层用100 mL乙酸乙酯萃取两次,合并有机相,用180 mL饱和碳酸氢钠溶液洗涤三次,120 mL饱和的氯化钠溶液洗涤两次,再用无水硫酸钠干燥,真空浓缩,除掉溶剂,得到油状的化合物2粗品8.99 g(产率91.4%)。粗产物无须纯化,直接用于下步反应。
2.2.42-吲哚乙酸乙酯(化合物3)的制备
将化合物2(4.00 g, 0.159 mol)溶解于50 mL丙酮溶夜,并转移至分液漏斗中,往其中加入400 mL的醋酸铵水溶液(4 mol/L)和120 mL 的TiCl3水溶液(15%,W/V),将混合物振摇7 min,形成深绿色溶液,再用400 mL乙醚萃取4次,合并有机相,分别用50 mL蒸馏水和50 mL的氯化钠溶液(10 %,W/V)洗涤,再用无水硫酸镁干燥。减压浓缩,剩余物除去有机溶剂,用石油醚—乙酸乙酯重结晶,得到白色结晶的目标化合物3 2.79 g(产率87.5%,纯度99.0%)。(HPLC归一化法:色谱柱Phecda C18柱(4.6 mm×150 mm, 3 μm);流动相0.02 mol/L磷酸二氢钾溶液(pH 6.0)—乙腈(65∶35);检测波长212 nm;柱温30 ℃;流速0.5 mL/min)。
2.2.5合成工艺条件研究
在目标化合物的合成中,我们首先用丙二酸二乙酯与KOH在无水乙醇中,在室温下搅拌下反应,TLC严格跟踪反应,在此条件下化合物1合成比较完全,得到较纯产品。第二步,在三氯化钛的的催化作用下,化合物2进行还原—缩合成环得目标化合物3,反应速度快,反应完全。第三步,2-硝基苯乙酸与化合物1进行亲核加成生成中间产物化合物2,反应条件不同,对产品的收率影响大。
在本研究中笔者主要从物料比n(2-硝基苯乙酸)∶n(化合物1)、反应温度和反应时间三个因素进行考虑,考察它对反应结果的影响。
3结果与讨论
3.1目标产物及中间产物的表征分析
3.1.1化合物2的表征结果及分析
粗产物用乙醇重结晶(结晶产率87%),得到浅褐色的粉末,m.p.54.8—57 ℃(理论熔点:56 ℃)(Found:C, 57.1; H, 5.1; N, 5.5. Calc. for C12H13NO5: C, 57.4; H, 5.2; N, 5.6%); νmax(Nujol)1 745 (ester C=O), 1 718(ketone C=O), 1 611,1 568,1 525 (aromatic NO2), 1 406, 1 339 (aromatic NO2), 1 305, 1 271, 1 203, 1 150, 1 050, and 1 027 cm-1; δH(400 MHz; CDCl3) 1.31(3 H, t, J 7 Hz, ethoxy CH3), 3.58 (2 H, s, CH2COOEt), 4.20 (2 H, q, J 7 Hz ethoxy, CH2), 4.21 (2 H, s, benzyl CH2), 7.28 (1 H, dd, J 8.6 1.6 Hz), 7.43 (1 H, td, J 8.6, 8.6, 1.6 Hz); 7.65 (1 H, td, J 8.6, 8.6, 1.6 Hz), and 8.06 (1 H, dd, J 8.6, 1.6 Hz); m/z 251.4 (M+, 1.4%), 163.9(21), 137.1(45), 120.09(53), 114.8(100), 87.3(32), 43.0(41), and 29.11(73)。检测结果证明所合成的物质为化合物2。
3.1.2目标化合物3的表征结果及分析
m.p.28—31 ℃; νmax(film)3 461 (NH), 1 725(C=O), 1 524,1 447,1 218, and 1 023 cm-1; δH(400 MHz; CDCl3) 1.49(3 H, t, J 7 Hz, ethoxy CH3), 3.72 (2 H, s, CH2COOEt), 4.20 (2 H, q, J 7 Hz ethoxy, CH2), 6.13 (1 H, s, 3-H), 7.07 (1 H, t, J 8 Hz), 7.13 (1 H, t, J 8 Hz); 7.34 (1 H, t, J 8 Hz), and 7.43(1 H, d, J 8 Hz), and 8.04(1H, br, NH); m/z 203.15 (M+, 45%) and 130.09(100)。测试数据表明所合成的物质即为目标产物,色谱纯度达98%。2-吲哚乙酸乙酯HNMR谱图如图2所示。
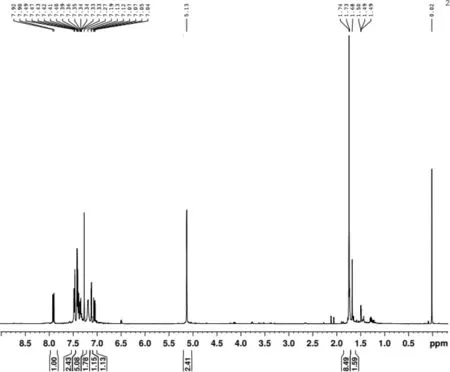
图2 2-吲哚乙酸乙酯 1HNMR谱图
3.2合成2-吲哚乙酸乙酯工艺的研究
3.2.1物料比(2-硝基苯乙酸∶化合物1)对产品收率的影响
实验条件:反应温度5 ℃,反应20 h,见表1。
表12-硝基苯乙酸:化合物1配比对反应结果的影响
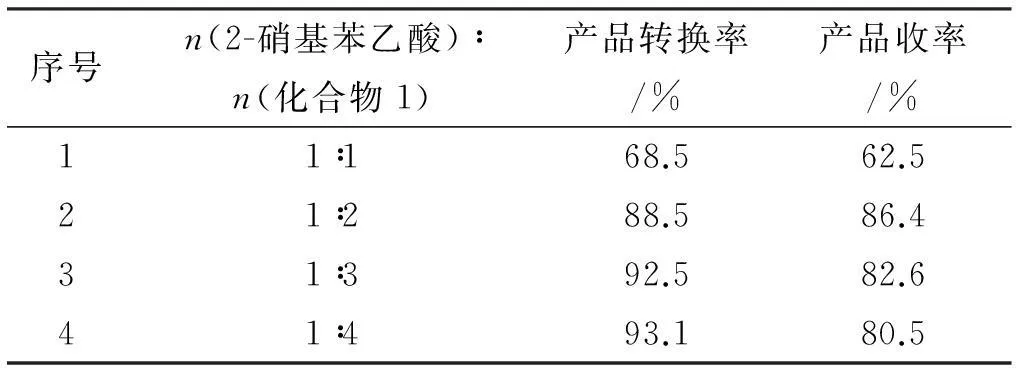
序号n(2-硝基苯乙酸)∶n(化合物1)产品转换率/%产品收率/%11∶168.562.521∶288.586.431∶392.582.641∶493.180.5
由表1所知,随着化合物1用量的增加,反应的转化率逐渐增大,当化合物1的用量过多时,可能是苯环上的取代反应增加,副产物增多,导致产品收率下降,而且也会影响后一步产物的纯度,也增加反应的成本。因此当n(2-硝基苯乙酸)∶n(化合物1)=1∶2时比较合适。
3.2.2反应温度对产品收率的影响
实验条件:物料比n(2-硝基苯乙酸)∶n(化合物1)=1∶2,反应时间20 h。
在N,N'-羰基二咪唑(CDI)作用下,为减少副反应的发生,应尽可能在低温下进行。由表2数据表明,反应温度为5 ℃时,反应效果最好,产品的收率达到最大。反应温度只要低于10 ℃,反应温度对该反应的影响不大。
表2温度对反应结果的影响
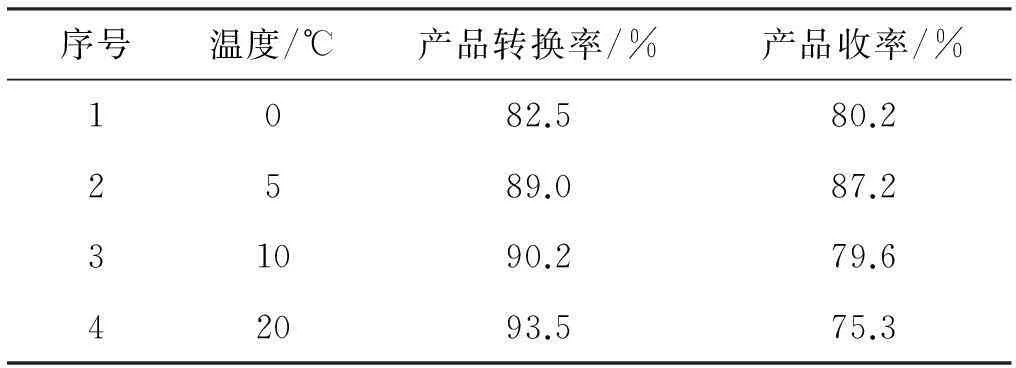
序号温度/℃产品转换率/%产品收率/%1082.580.22589.087.231090.279.642093.575.3
3.2.2反应时间对产品收率的影响
实验条件:物料比n(2-硝基苯乙酸)∶n(化合物1)=1∶2,反应温度5 ℃。
表3数据表明,产品转化率随反应时间的增加而增大,但反应时间过长降低主反应的选择性,副反应增多,使收率下降。故反应时间在20 h左右比较合适。
表3反应时间对反应结果的影响
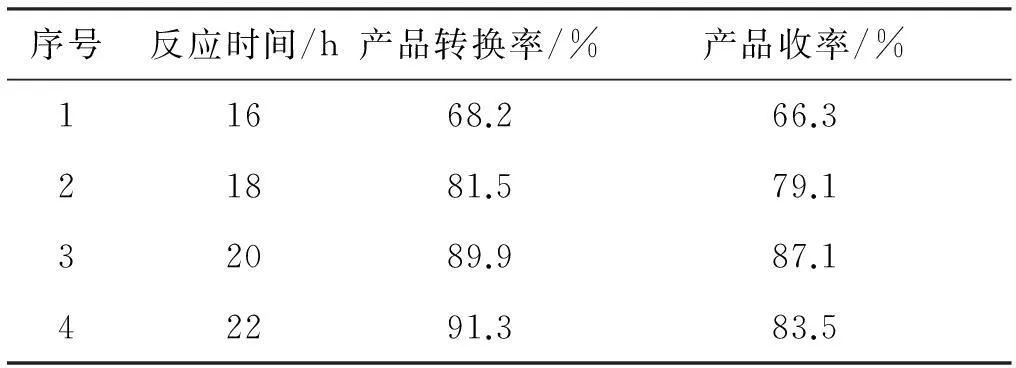
序号反应时间/h产品转换率/%产品收率/%11668.266.321881.579.132089.987.142291.383.5
3.2.3合成工艺的正交实验设计
综上结论,根据确定的几个影响因素进行了正交实验设计,结果如表4和表5所示.
表42-吲哚乙酸乙酯合成影响因子水平表

水平/因子123A∶n(2-硝基苯乙酸)∶n(化合物11∶11∶21∶3B∶反应温度/℃0510D∶反应时间/h161820
表5正交实验及实验结果
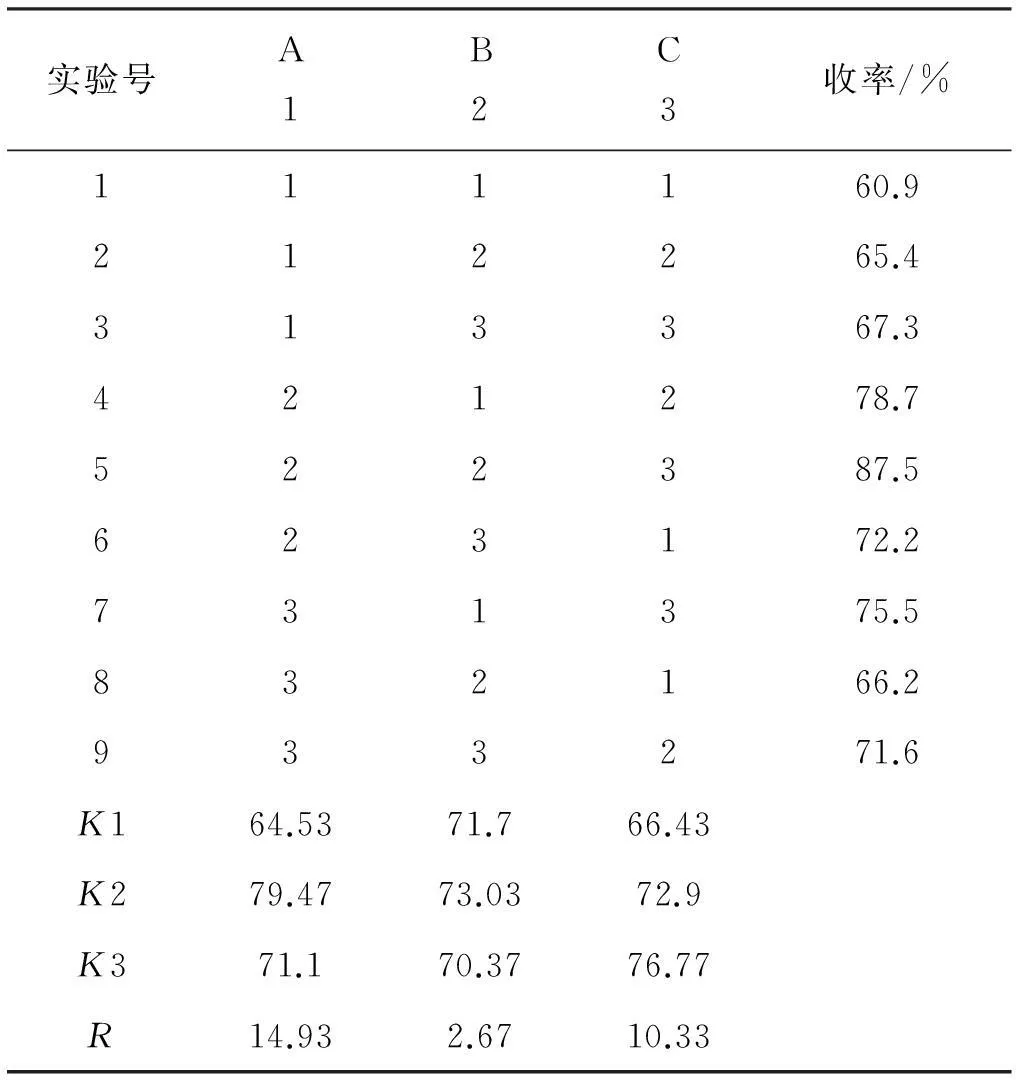
实验号A1B2C3收率/%111160.9212265.4313367.3421278.7522387.5623172.2731375.5832166.2933271.6K164.5371.766.43K279.4773.0372.9K371.170.3776.77R14.932.6710.33
从正交表中的极差数据分析中可以看出对2-吲哚乙酸乙酯合成影响最大的因素是物料配比(因素A)、其次是反应时间(因素C)及反应温度(因素B)。从极差(R)数据分析可知最佳的合成条件为A2B2C3,即物料配比n(2-硝基苯乙酸)∶n(化合物1)为1∶2,反应温度5 ℃,反应时间为20 h。
在最佳合成工艺条件下,重复上述实验,数据如表6所示。
表6最佳工艺条件稳定性实验
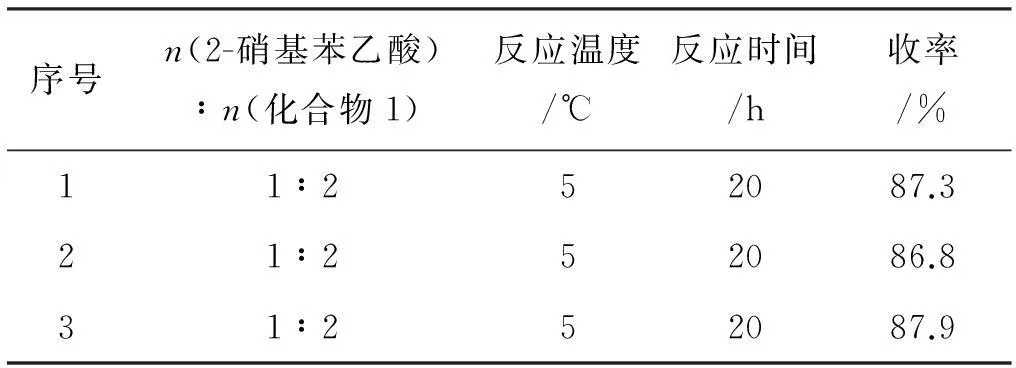
序号n(2-硝基苯乙酸)∶n(化合物1)反应温度/℃反应时间/h收率/%11∶252087.321∶252086.831∶252087.9
实验证明,该合成工艺条件稳定,条件温和,总收率高。
4结论
4.1本合成路线无须将2-硝基苯乙酸酰基化,利用强反应活性物质N,N' -羰基二咪唑,在其作用下,通过2-硝基苯乙酸与化合物1的亲核加成即可得到中间体产物化合物2,分离后粗产物不用纯化,可直接在三氯化钛的水合丙酮溶液中进行还原环化即得到目标产物2-吲哚乙酸乙酯。
4.2最佳合成工艺为物料配比n(2-硝基苯乙酸)∶n(化合物1)为1∶2,反应温度5 ℃,反应时间为20 h,此条件下收率为87.5%。
4.3合成的最终产物为浅棕色固体,熔点约为28—31 ℃,经IR、1HNMR和MS表征,确认为目标产物2-吲哚乙酸乙酯。
4.4合成的最佳工艺条件重复性实验稳定,合成反应在0—40 ℃条件下均可进行,20 h内即可完成目标产物3的合成,且中间体产物无须分离,此外,该反应原料易得,反应只需3步,步骤和成本等方面则大大缩减。
参考文献:
[1]Cacchi S, Fabrizi G. Synthesis and functionalization of indoles through palladium-catalyzed reactions [J]. Chemical reviews, 2005, 105(7): 2873-2920.
[2]Simoneau C A, Strohl A M, Anem B G. One-pot synthesis of polysubstituted indoles from aliphatic nitro compounds under mild conditions [J]. Tetrahedron Letters, 2007, 48(10): 1809-1811.
[3]Nishiyama Y, Naitoh Y, Sonoda N. A new synthetic method of 1,4-dihydro-2H-3,1-benzoxazin-2-ones:selenium-catalyzed reductive carbonylation of aromatic nitro compounds with carbon m onoxide [J]. Synlett, 2004, 1(5): 886-888.
[4]徐小军, 尤庆亮, 余朋高, 等. Fischer法合成2,5-二甲基吲哚的工艺研究 [J]. 化学与生物工程, 2013, 30(4): 59-62.
[5]官海伟, 解正峰. 合成双吲哚甲烷类衍生物的研究进展 [J]. 有机化学, 2012, 32(7): 1195-1207.
[6]周峰, 郑灿辉, 朱驹, 等. 取代-1,3-二氢吲哚-2-酮化合物的合成 [J]. 中国医药工业杂志, 2013, 44(7): 660-662.
[7]郭红云, 田金金. 碱性离子液体催化下一锅三组分合成螺环吲哚衍生物 [J]. 有机化学, 2011, 31(5):752-756.
[8]Wenkert E, Marsaioli A J, Moeller PDR. Formal total synthesis of rosefuran and eldanolide [J]. Journal of Chromatography, 1988, 25(440): 449-453.
[9]翟帆, 麻纪斌, 胡碧荣. 6-溴-2-溴甲基-5-羟基-1-甲基吲哚-3-羧酸乙酯的合成工艺研究 [J]. 应用化工, 2013, 42(6):1068-1051.
[10]Attia M I, Güclü D, Hertlein B, et al. Synthesis, NMR conformational analysis and pharm- acological evaluation of 7,7a,13,14-tetrahydro-6H-cyclobuta [b] pyrimido[1,2-a:3,4-a’] di-indole analogues as melaonin receptor ligands [J]. Organic & Biomolecular Chemistry, 2007, 5(13): 2129-2137.
[11]Moody C J, Rahimtoola K F. Diels-Alder Reactivity of Pyrano [4,3-b] indol-3-ones, Indole 2,3-Quinodimethane Analogues [J]. Journal of the Chemical Society, Perkin Transactions1990, 2(35): 673-679.
