酪氨酸猝灭Eosin Y的荧光
2015-12-05王经东于安池
王经东 李 爽 吕 荣 于安池
(中国人民大学化学系, 北京 100872)
酪氨酸猝灭Eosin Y的荧光
王经东 李 爽 吕 荣 于安池*
(中国人民大学化学系, 北京 100872)
氨基酸残基对探针分子的荧光猝灭行为可以为生物大分子的结构及构象动力学研究提供重要的信息.本文运用飞秒瞬态吸收光谱和时间相关单光子计数实验系统研究了在水(H2O)和氘代水(D2O)溶液中乙酰基取代酪氨酸(AcTyr)对Eosin Y的超快荧光猝灭动力学过程. 发现导致AcTyr对Eosin Y荧光猝灭的主要原因是由于它们之间形成了短寿命的基态复合物. 我们还发现Eosin Y与AcTyr形成的基态复合物的激发态寿命具有明显的动力学同位素效应, 表明AcTyr对Eosin Y的荧光猝灭是通过质子耦合电子转移过程发生的.
飞秒瞬态吸收光谱; 时间相关单光子计数; 基态复合物; 酪氨酸猝灭; 质子耦合电子转移
1 Introduction
Quenching of a fluorescent probe by amino acid residues can provide valuable information about structures and conformational dynamics of peptides and proteins.1–11It is known that many processes such as fluorescence resonance energy transfer (FRET), Dexter electron exchange, exciplex or ground-state complex formation, and photoinduced electron transfer (PET), can lead to the fluorescence quenching of a fluorophore.12Unlike FRET and Dexter transfer processes, PET does not require the spectral overlap between a donor and an acceptor and is only governed by the redox chemistry of the donor and acceptor. Besides, PET requires a contact formation between a donor and an acceptor at a sub-nanometer scale and makes it an elegant method to probe subtle structural or conformational changes in biopolymers.13
Among amino acids, tryptophan and tyrosine are the two that can cause the most obvious fluorescence quenching of a fluorophore in aqueous solution or in protein.1–11,13–30Up to date, thefluorescence quenching of a fluorophore by tryptophan has been extensively studied and it is generally reported that tryptophan quenches the fluorescence of the fluorophore via a PET interaction.4–7,10,11,14–26However for the fluorescence quenching of a fluorophore by tyrosine, its mechanism still debates.14–17,28–30Some studies suggest that tyrosine quenches the fluorescence of a fluorophore via a PET process,14–17while some other studies suggest that tyrosine quenches the fluorescence of a fluorophore via a photoinduced proton-coupled electron transfer (PCET) process.28–30It is known that both PET and PCET are important elementary steps in biochemical reactions.31–33
Recently, we studied the ultrafast fluorescence quenching dynamics of Atto655 in N-acetyl-tyrosine (AcTyr) solution with femtosecond transient absorption spectroscopy and found that AcTyr quenched the fluorescence of Atto655 in aqueous solution via a PCET interaction.34To search another PCET system, we studied the fluorescence quenching dynamics of Eosin Y by AcTyr in aqueous solution in this work. The reason that we selected Eosin Y is due to that the photophysics and photochemistry of Eosin Y have been of widespread interest and it is frequently used as a fluorescent probe and biological stain.35Due to the low solubility of tyrosine in water, we selected AcTyr instead of tyrosine. In this work, we first obtained the transient absorption spectra of Eosin Y in the absence and presence of 200 mmolL–1AcTyr solution at various delay times with a femtosecond transient absorption spectrometer. Then, we measured the first electronic excited-state decays of Eosin Y in the presence of different concentration of AcTyr in H2O and D2O solutions. We found that the decay time of the first electronic excited-state of Eosin Y in the presence of AcTyr in D2O solution was different with that in H2O solution. The molecular structures of both Eosin Y and N-acetyl-tyrosine are displayed in Scheme 1.
2 Materials and methods
Eosin Y (> 99%) and N-acetyl-tyrosine (> 99%) were purchased from Sigma-Aldrich and used as received. 1 × TE (Tris-HCl EDTA) buffer (10 mmolL–1Tris-HCl + 1 mmolL–1EDTA, pH = 8.0) was diluted from 100 × TE buffer (Sigma-Aldrich). Ultrapure H2O (18.2 MΩcm) was obtained through a Millipore Milli-Q water purification system. Ultra-D D2O (> 99.9%) was purchased from Sigma-Aldrich and used as received.
Steady-state absorption spectra were recorded on a Varian Cary 50 UV-Vis spectrometer. Steady-state fluorescence spectra were recorded on a Perkin-Elmer LS-55 luminescence spectrometer. Fluorescence lifetimes were obtained on a homemade time-correlated single-photon counting (TCSPC) apparatus.36Briefly, the output of an optical parametric amplifier (OPA) pumped by a Spectra Physics 1 kHz amplified Ti:Sapphire laser was used as the excitation light. The emission was collected and sent into a Princeton Instruments SP2358 monochromator and detected with a PDM-50CT single photon avalanche diode. The NIM-output of the PDM 50CT and the output of a fast TDA 200 photodiode were respectively connected to a PicoQuant GmbH TimeHarp 200 correlator as the start and stop pulses. Magic-angle detection was used. The instrumental response function of this equipment was about 180 ps.
The femtosecond transient absorption setup was described elsewhere.37Briefly, the outputs of a Spectra Physics 1 kHz amplified Ti:sapphire laser were used to pump an OPA and to generate the white light continuum, respectively. The outputs of the OPA were used as the pump pulses, and the white light continuum generated by a spinning fused silica disk were used as the probe pulses. The timing between the pump and probe pulses was controlled using a Newport M-ILS250CC motorized translation stage. The time resolution of this apparatus was about 150 fs.
Scheme 1 Molecular structures of Eosin Y and N-acetyl-tyrosine
In the TCSPC measurements, the concentrations of the samples were kept at about 1 × 10–6molL–1. In the femtosecond transient absorption measurements, the concentrations of the samples were kept at about 1 × 10–5molL–1. A homemade magnet stirring bar was placed inside a 1 mm path length sample cell and rotated by an external magnet motor to keep the sample solution fresh.
3 Results and discussion
The formation of a ground-state complex between a fluorophore and a quencher can be revealed through its ensemble steady-state absorption spectra.16–18Fig.1 displays the steadystate absorption spectra of Eosin Y in the presence of different concentrations of AcTyr in 1 × TE pH = 8.0 buffer. The inset in Fig.1 is the expended view of the steady-state absorption spectra of Eosin Y with different concentrations of AcTyr. Clearly, the absorbance of Eosin Y decreases slightly and its absorption maximum shifts bathochromically upon addition of AcTyr. Furthermore, titration of Eosin Y with AcTyr solution shows an isosbestic point at around 522 nm in the absorption spectra of Eosin Y with different concentrations of AcTyr (inset of Fig.1). These findings demonstrate that a ground-state complex between Eosin Y and AcTyr was formed in aqueous solution.
To explore the interaction between Eosin Y and AcTyr, we respectively recorded the fluorescence spectra of Eosin Y under different concentrations of AcTyr in aqueous solution. Fig.2 shows the absorbance-corrected fluorescence spectra of Eosin Y in the presence of different concentrations of AcTyr in 1 × TE pH = 8.0 buffer. The fluorescence intensity of Eosin Y de-creases upon addition of AcTyr, indicating that AcTyr can quench the fluorescence of Eosin Y.
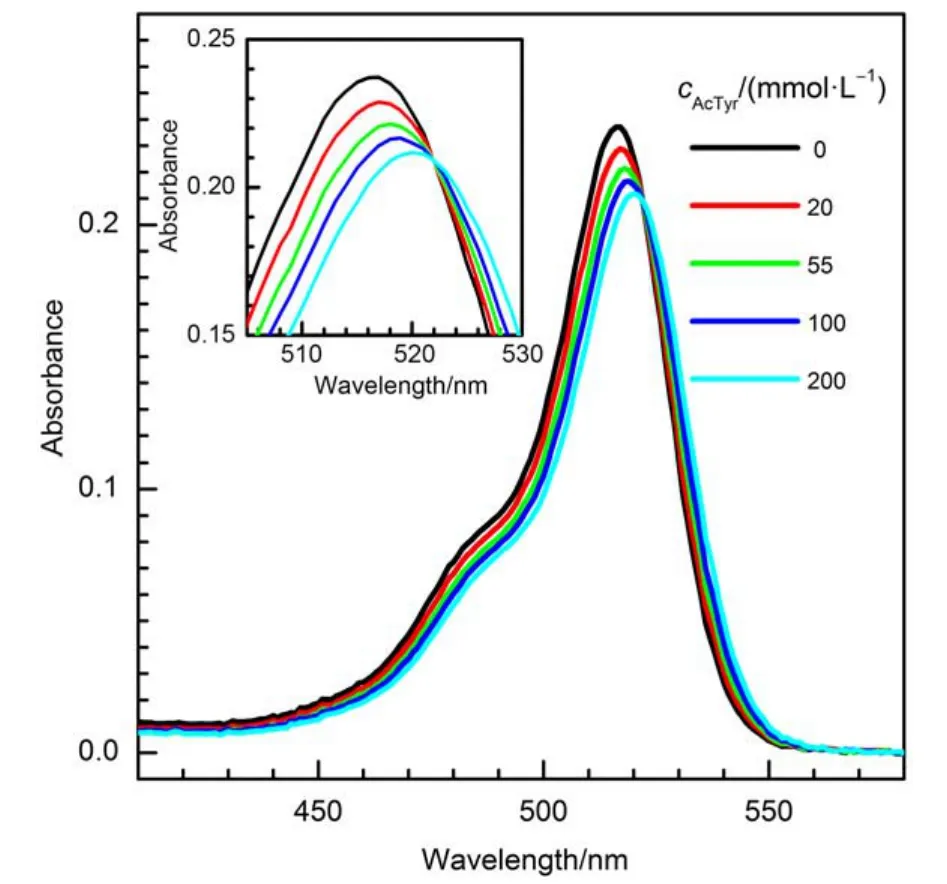
Fig.1 Steady-state absorption spectra of Eosin Y in the presence of different concentrations of AcTyr in 1 × TE pH = 8.0 buffer
To quantitatively analyze the fluorescence quenching behavior of Eosin Y by AcTyr, we displayed the steady-state fluorescence intensity Stern-Volmer (SV) plot of Eosin Y in the presence of AcTyr in 1 × TE pH = 8.0 buffer in Fig.3. It is clear that the SV plot of Eosin Y in AcTyr solution has an upward curvature. Through a quadratic Stern-Volmer model F0/F = (1 + KS[Q])(1 + KD[Q])(F and F0are the fluorescence intensity of Eosin Y in the presence and absence of AcTyr, and [Q] is the concentration of AcTyr), which incorporated both static (KS) and dynamic (KD) components, we determined that AcTyr quenches the fluorescence of Eosin Y with a KSvalue of (14.5 ± 1.0) mol–1L and a KDvalue of (3.0 ± 0.6) mol–1L in 1 × TE pH = 8.0 buffer solution. The KSvalue is about 5 times larger than the KDvalue, indicating that the static fluorescence quenching between Eosin Y and AcTyr through a ground-state complex formation is dominant in the fluorescence quenching of Eosin Y by AcTyr, which is in agreement with our steadystate absorption observation (Fig.1).
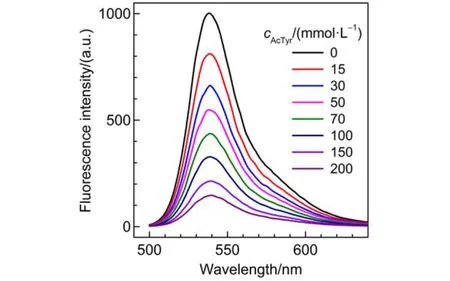
Fig.2 Absorbance-corrected steady-state fluorescence spectra of Eosin Y in the presence of different concentrations of AcTyr in 1 × TE pH = 8.0 buffer
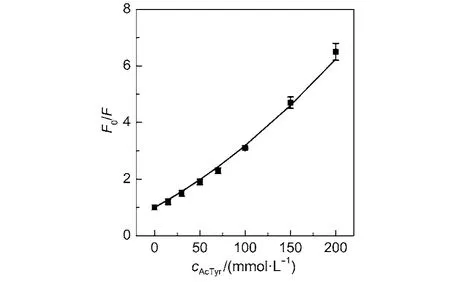
Fig.3 Steady-state Stern-Volmer plot of Eosin Y in the presence of AcTyr in 1 × TE pH = 8.0 buffer
To confirm that the dynamic quenching process between Eosin Y and AcTyr is minimal, we also recorded the fluorescence decay kinetics of Eosin Y in the presence of different concentrations of AcTyr in aqueous solution by means of TCSPC experiments (The time resolution of our TCSPC setup is low and it can only catch the dynamic quenching process). Fig.4 shows the magic-angle fluorescence decay curves of Eosin Y under different concentrations of AcTyr in 1 × TE pH = 8.0 buffer solution. The fluorescence decay of Eosin Y becomes slightly faster upon increasing AcTyr concentration. The fluorescence decay of Eosin Y without AcTyr can be fitted by a single exponential decay function, while the fluorescence decays of Eosin Y with AcTyr need either a single exponential decay function or a summation of two exponential decay functions to be fitted. Table 1 summarizes all fitting parameters for the magic-angle fluorescence decays of Eosin Y in the presence of different concentrations of AcTyr in 1 × TE pH = 8.0buffer solution. escence quenching modelτ and τ0are the fluorescence lifetime of Eosin Y in the presence and absence of AcTyr), we derived that AcTyr quenches dynamically the fluorescence of Eosin Y with amol–1L in 1 × TE pH = 8.0 buffer. The obtainedvalue ((2.2 ± 0.4) mol–1L) with time-resolved data shown in Fig.5 is quite similar as the obtained KDvalue ((3.0 ± 0.6) mol–1L) with steadystate data shown in Fig.3. With the obtainedvalue ((2.2 ± 0.4) mol–1L) and fluorescence lifetime of Eosin Y (1.10 ns), we derived a collision quenching rate constant, kD= (2.0 ± 0.3) × 109mol–1Ls–1, for the bimolecular fluorescence quenching
With the averaged lifetime (< τ >) listed in Table 1, we obtained the time-resolved Stern-Volmer plot of Eosin Y in the presence of AcTyr in aqueous solution (Fig.5). Obviously, the obtained quenching efficiency of Eosin Y by AcTyr at each AcTyr concentration with the time-resolved fluorescence measurement (Fig.5) is extremely smaller than that with the steadystate fluorescence measurement (Fig.3). Through a linear fluor-process between Eosin Y and AcTyr. The obtained kDvalue is about 4 times smaller than the bimolecular collision rate constant predicted by the Smoluchowski equation for a diffusionlimited process,38indicating that not all collisions between Eosin Y and AcTyr are efficient to quench the fluorescence of Eosin Y.
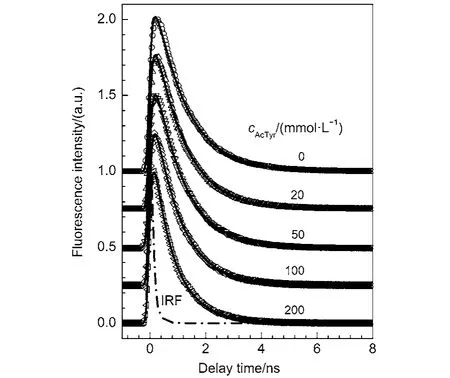
Fig.4 Magic-angle fluorescence decays of Eosin Y in the presence of different concentrations of AcTyr in 1 × TE pH = 8.0 buffer
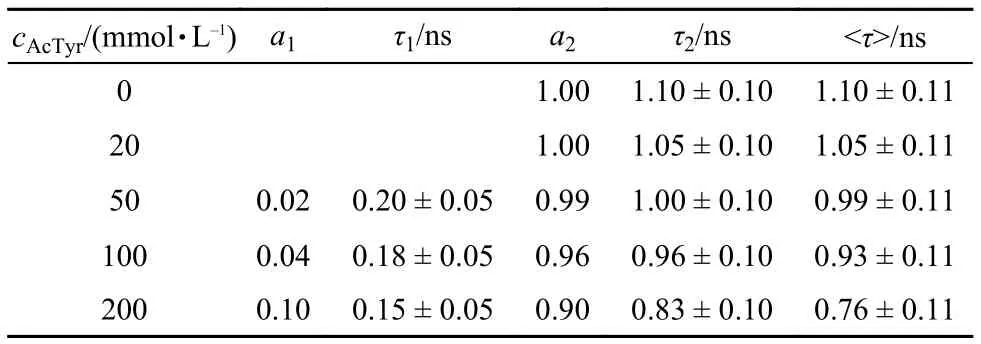
Tabl1 Fluorescence decay parameters for Eosin Y in the presence of different concentrations of AcTyr in 1 × TE pH = 8.0 buffer, obtained from magic-angle time-correlated single-photon counting measurements
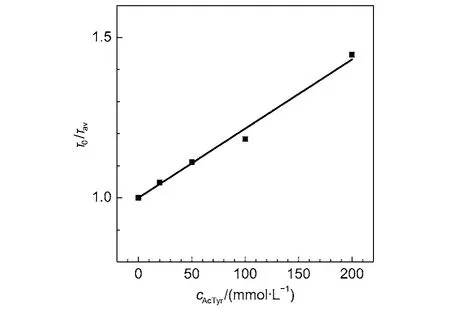
Fig.5 Time-resolved Stern-Volmer plot of Eosin Y in the presence ofAcTyr in 1 × TE pH = 8.0 buffer
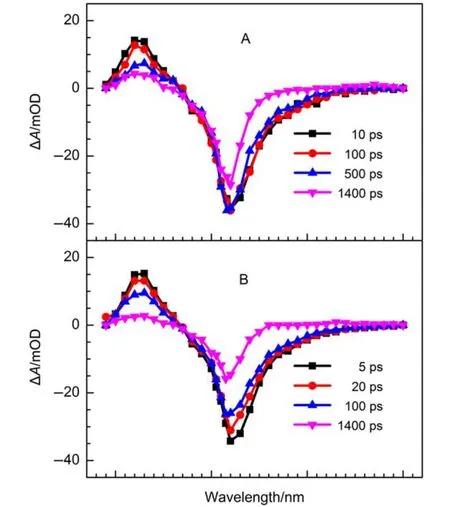
Fig.6 Femtosecond transient absorption spectra of Eosin Y in the absence (A) and presence (B) of 200 mmolL–1AcTyr solution in 1 × TE pH = 8.0 buffer at various time delays
To investigate the ultrafast fluorescence quenching dynamics of Eosin Y by AcTyr, we measured the transient absorption spectra of Eosin Y in the absence and presence of 200 mmolL–1AcTyr solution at various time delays by using a femtosecond transient absorption spectrometer, as shown in Fig.6. The obtained transient absorption spectra of eosin Y in aqueous solution at various delays (Fig.6A) are in agreement with the previous report.39The transient absorption spectra of Eosin Y mainly consists of two bands: one absorption band located in the wavelength range of 390–470 nm due to its singlet excited-state absorption and one bleaching band located in the wavelength range of 470–650 nm where is coinciding with its ground-state absorption and excited-state stimulated emission. Due to the limitation scanning range of our femtosecond transient absorption spectrometer and the relatively low absorption extinction coefficient of the triplet electronic state of Eosin Y, we only observed a tiny absorption band in the wavelength range of 600–700 nm arising from its triplet electronic state at the longer delay time (i.e., 1400 ps). Besides, it is also found that the time evolution of the transient absorption spectra of Eosin Y in 200 mmolL–1AcTyr solution is faster than that in aqueous solution.
Since the time evolution of the transient absorption spectra of Eosin Y in the wavelength range of 390–470 nm (first electronic excited-state absorption) is much simpler than that in the wavelength range of 470–700 nm, we monitor the first electronic excited-state decay kinetics of Eosin Y to catch its fluores-cence quenching dynamics by AcTyr in aqueous solution. Fig.7 displays the first electronic excited-state decay kinetics of Eosin Y in the presence of different concentrations of AcTyr in 1 × TE pH = 8.0 buffer solution. Clearly, the decay of the first electronic excited-state of Eosin Y becomes faster upon addition of AcTyr. The decay of the first electronic excited-state of Eosin Y without AcTyr can be fitted by a single exponential decay function, while the decays of the first electronic excited-state of Eosin Y with AcTyr need a summation of two exponential decay functions to be fitted. All fitting parameters are summarized in Table 2. From the data listed in Table 2, it is found that the time constant of the fast component (τ1) of Eosin Y in the presence of AcTyr does not vary with the increase of AcTyr concentration, but its amplitude (a1) does. This is a well-known behavior40and indicates the formation of a ground-state complex between Eosin Y and AcTyr in aqueous solution, which is also consistent with our steady-state absorption and fluorescence spectroscopy measurements. Besides, the slower time constant of Eosin Y in different AcTyr concentrations (τ2listed in Table 2) also agrees well with the obtained averaged time constant from our TCSPC measurement (< τ > listed in Table 1), which reflects the dynamic fluorescence quenching process between Eosin Y and AcTyr.
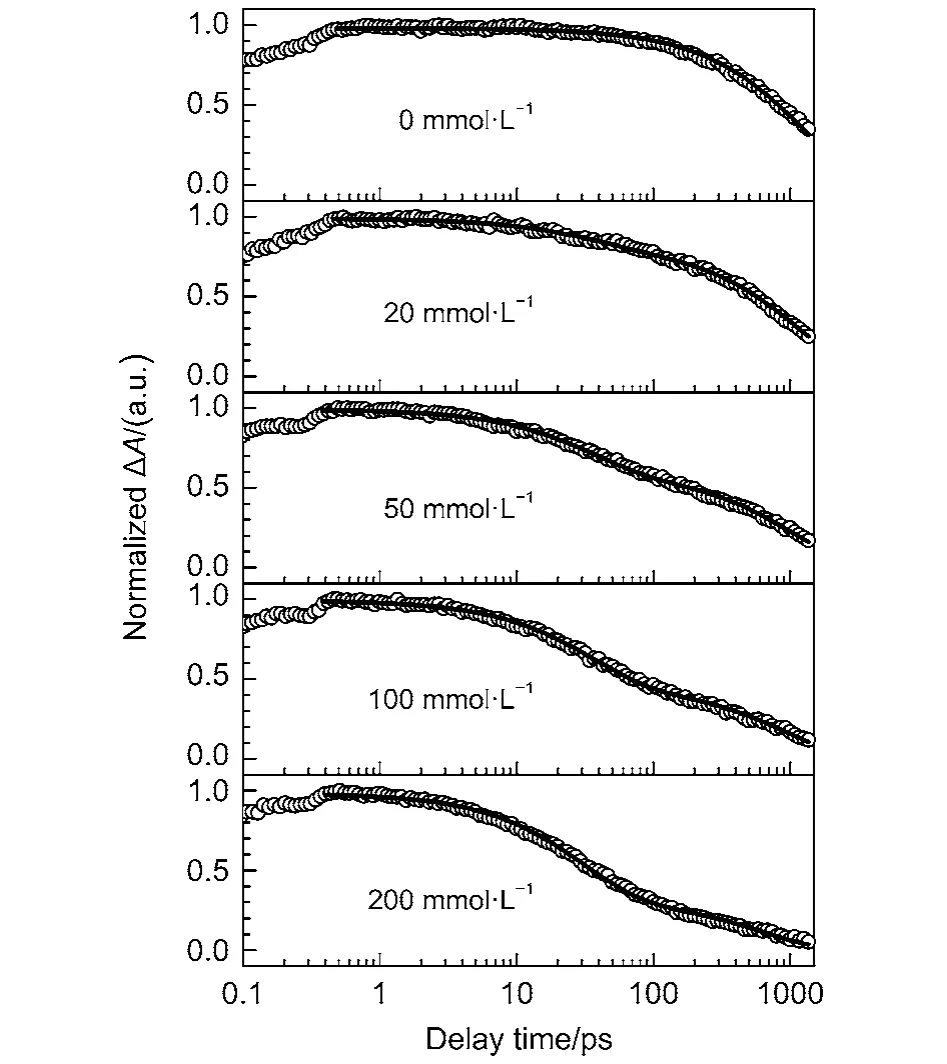
Fig.7 Magic-angle femtosecond pump-probe transients of Eosin Y in the presence of different concentrations of AcTyr in 1 × TE pH = 8.0 H2O buffer
Researchers generally use so-called “static” and “dynamic”quenching to account for the deactivation of excited fluorophores.18,41However, Zewail et al.40pointed out that this distinction depends on the actual timescale of the utilized experimental methods. Herein, we measured the fluorescence quenching kinetics of Eosin Y by AcTyr with a femtosecond transient absorption spectrometer. The time resolution of the femtosecond transient absorption spectrometer (150 fs ) is far higher than that of typical time correlated single photon counting setup (100 ps). Thus, the ~35 ps component listed in the Table 2 should be the excited-state lifetime of the ground-state complex formed between Eosin Y and AcTyr in 1 × TE H2O buffer solution.
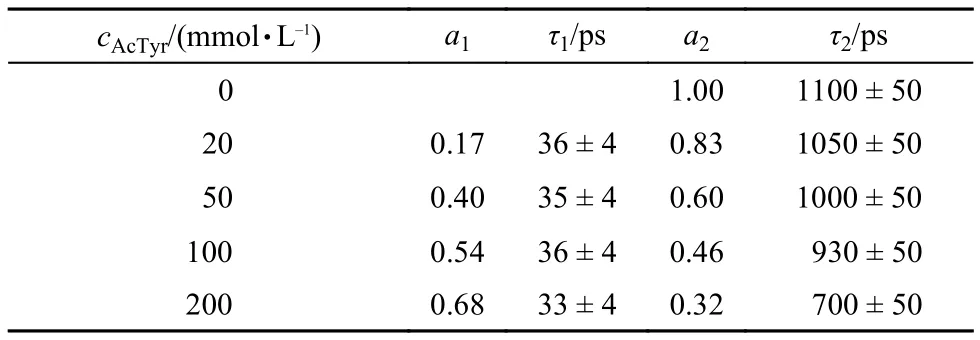
Tabl2 Excited-state decay parameters for Eosin Y in the presence of different concentrations of AcTyr in 1 × TE pH = 8.0 H2O buffer, obtained from femtosecond transient absorption measurements
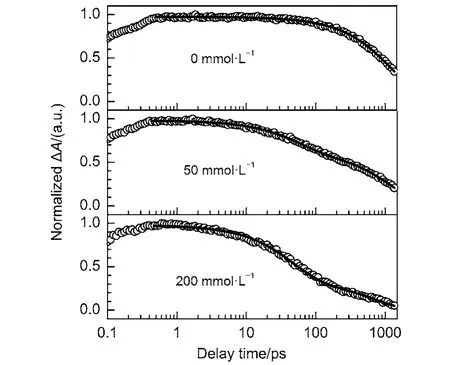
Fig.8 Magic-angle femtosecond pump-probe transients of Eosin Y in the presence of different concentrations of AcTyr in 1 × TE pH = 8.0 D2O buffer
Recently, it is found that AcTyr quenches the fluorescence of Atto655 via a PCET process in aqueous solution.34Moreover, it is reported that the observation of a kinetic isotope effect (KIE) is the hallmark of a PCET reaction.33,42,43To confirm whether the fluorescence quenching of Eosin Y by AcTyr in aqueous solution undergoes a PCET process or not, we also measured the first electronic excited-state decays of Eosin Y in the presence of different concentrations of AcTyr in 1 × TE D2O buffer solution, as shown in Fig.8. Table 3 listed the fitting parameters for the excited-state decays of Eosin Y in the presence of different concentrations of AcTyr in 1×TE D2O buffer solution. Similarly, the time constant of the fast component (~54 ps) of Eosin Y in the presence of AcTyr in 1 × TE D2O buffer solution also does not vary with the increase of AcTyr concentration, but its amplitude does. Thus, the ~54 ps component listed in the Table 3 is also the excited-state lifetime of the ground-state complexformed between Eosin Y and AcTyr in 1 × TE D2O buffer solution. The excited-state lifetime of the ground-state complex formed between Eosin Y and AcTyr in 1 × TE D2O buffer solution is different with that in 1 × TE H2O buffer solution and it shows an obvious KIE effect (~54 ps in 1 × TE D2O buffer solution vs ~35 ps in 1 × TE H2O buffer solution), indicating that AcTyr also quenches the fluorescence of Eosin Y via a PCET process.
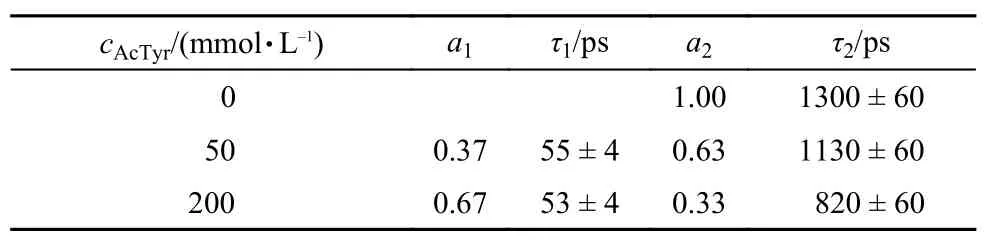
Tabl3 Excited-state decay parameters for Eosin Y in the presence of different concentrations of AcTyr in 1×TE D2O buffer, obtained from femtosecond transient absorption measurements
In this work, we studied the fluorescence quenching dynamics of Eosin Y by AcTyr in aqueous solution and found that the excited-state lifetime of the ground-state complex formed between Eosin Y and AcTyr in 1 × TE H2O buffer solution is about 35 ps. With the kinetic scheme shown in our previous reports,26we derived that the PCET rate from AcTyr to Eosin Y is about 2.8 × 1010s–1. For a PCET process, various levels of theory have been employed.32,33However in the recent, Mayer and coworkers44,45proved that the PCET reaction can be well described by the semiclassical Marcus electron transfer equation31
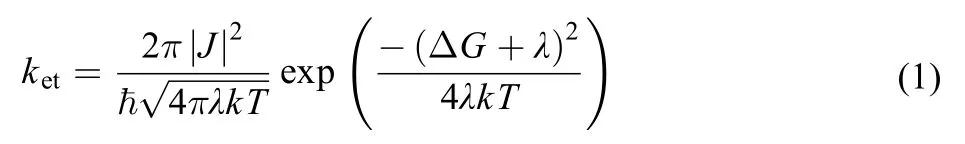
where ΔG is the driving force for the electron transfer reaction, J is the electronic coupling matrix element between donor and acceptor, λ is the reorganization energy, ħis the Planck constant, k is the Boltzman constant, and T is the temperature. The driving force ΔG can be estimated by using Weller’s equation46

where Eoxis the first one-electron oxidation peak potential of the donor, Eredis the first one-electron reduction peak potential of the acceptor, ESis the energy of the zero-zero transition to the lowest excited singlet state, and C is the solvent dependent Coulombic interaction energy, which can be neglected in moderately polar environment.
From literature, it is found that the peak potential for oneelectron reduction of Eosin Y is around –0.79 V (vs normal hydrogen electrode, NHE)47and the first one-electron oxidation peak potential of AcTyr in aqueous solution is around 0.89 V (vs NHE)29,48. Besides, from the steady-state absorption and fluorescence spectra shown in Figs.1 and 2, it is found that the zero-zero transition energy (ES) of Eosin Y in aqueous solution to be 2.35 eV (~527 nm). Substituting these values into equation (2), we derived that ΔG = –0.67 eV for the PCET reaction between Eosin Y and AcTyr. Assuming a typical reorganization energy of λ = 1.2 eV for aqueous solution40,45,49and substituting the PCET rate (2.8 × 1010s–1) from AcTyr to Eosin Y into equation (1), we derive that the electronic coupling constant for the PCET reaction between Eosin Y and AcTyr is about 35 cm–1. Gotz et al.15reported that the electronic coupling constant for the reaction between labeled fluorescein and Tyr residue in FluA-Fl complex is 140 cm–1, which is quite different with our obtained electronic coupling constant for the reaction between Eosin Y and AcTyr. Interestingly, Mayer and coworkers45studied the PCET reactions of a series of hydrogen-bonded phenols and found that the electronic coupling constants for the reactions of hydrogen-bonded phenols are in the range of 20–30 cm–1, which is quite similar to our obtained electronic coupling constant for the reaction between Eosin Y and AcTyr.
The fluorophore-tryptophan pairs have been widely used to monitor the conformational dynamics of peptides and proteins.4–8,13The current study demonstrates that the fluorophore-tyrosine pairs could also be employed to study the conformational dynamics of peptides and proteins. Fluorescence correlation spectroscopy (FCS)50has proved to be one of important experimental methods to study the conformational dynamics of a biopolymer. Under the assumption that the brightness (Q) of the dark species is zero, researchers could extract both the forward and reverse rate constants from a single FCS experimental curve,18which are important parameters to understand the conformational dynamics of a biopolymer. However, our recent study shows that the assumption of Q = 0 would introduce noticeable errors in its equilibrium constant for the systems with Q ≥ 0.01.34The value of Q for the dark species has to be determined with an additional experiment so as to obtain the correct forward and reverse rate constants. With the data listed in Table 2, we derived that the relative brightness of the groundstate complex formed between Eosin Y and AcTyr (Q = τ1/τF, where τFis the fluorescence lifetime of free dye) is 0.032 ± 0.004. Suppose a reaction (K = 1) involving Eosin Y and tyrosine in biopolymers, it will introduce about –12% deviation of its equilibrium constant under the assumption of Q = 0. The femtosecond study on the ultrafast fluorescence quenching dynamics of a fluorophore by amino acid residue in aqueous solution or in protein can provide valuable parameter (Q) for the study of the conformational dynamics of a biopolymer with the FCS method.
4 Conclusions
We studied the ultrafast fluorescence quenching dynamics of Eosin Y in the presence of AcTyr in H2O and D2O solutions with steady-state absorption and fluorescence spectroscopy, time-resolved fluorescence spectroscopy, and femtosecond transient absorption spectroscopy. Both the steady-state and time-resolved studies demonstrate that the formation of a ground-state complex between Eosin Y and AcTyr is the major process to cause the fluorescence quenching of Eosin Y by AcTyr in aqueous solution. The kinetic isotope effect on the first electronic excited-state decay kinetics of Eosin Y in the presence of AcTyr reveals that AcTyr quenches the fluorescence of Eosin Y in aqueous solution via a PCET process. With the version of the semiclassical Marcus electron transfer theory,we derived that the electronic coupling constant for the PCET reaction between Eosin Y and AcTyr in aqueous solution is around 35 cm–1. The obtained electronic coupling constant for the reaction between Eosin Y and AcTyr does not agree with the repoted electronic coupling constant for the reaction between labeled fluorescein and Tyr residue in FluA-Fl complex by Gotz et al.,15but it agrees well with the electronic coupling constants for the reactions of hydrogen-bonded phenols by Mayer and coworkers.45With the obtained kinetic data, we derived that the relative brightness of the formed ground-state complex between Eosin Y and AcTyr is 0.032 ± 0.004, which is an important parameter to understand the conformational dynamics of a biopolymer.
(1)Michalet, X.; Weiss, S.; Jager, M. Chem. Rev. 2006, 106, 1785. doi: 10.1021/cr0404343
(2)Royer, C. A. Chem. Rev. 2006, 106, 1769. doi: 10.1021/cr0404390
(3)Edman, L.; Mets, U.; Rigler, R. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 6710. doi: 10.1073/pnas.93.13.6710
(4)Neuweiler, H.; Banachewicz, W.; Fersht, A. R. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 22106. doi: 10.1073/pnas.1011666107
(5)Neuweiler, H.; Doose, S.; Sauer, M. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 16650. doi: 10.1073/pnas.0507351102
(6)Rogers, J. M. G.; Poishchuk, A. L.; Guo, L.; Wang, J.; DeGrado, W. F.; Gai, F. Langmuir 2011, 27, 3815. doi: 10.1021/la200480d
(7)Chen, H.; Rhoades, E.; Butler, J. S.; Loh, S. N.; Webb, W. W. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 10459. doi: 10.1073/pnas.0704073104
(8)Doose, S.; Neuweiler, H.; Barsch, H.; Sauer, M. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 17400. doi: 10.1073/pnas.0705605104
(9)Yang, H.; Luo, G. B.; Karnchanaphanurach, P.; Louie, T. M.; Rech, I.; Cova, S.; Xun, L. Y.; Xie, X. S. Science 2003, 302, 262. doi: 10.1126/science.1086911
(10)Hudgins, R. R.; Huang, F.; Gramlich, G.; Nau, W. M. J. Am. Chem. Soc. 2002, 124, 556. doi: 10.1021/ja010493n
(11)Marme, N.; Knemeyer, J. P.; Wolfrum, J.; Sauer, M. Angew. Chem. Int. Edit. 2004, 43, 3798.
(12)Goldberg, J. M.; Batjargal, S.; Chen, B. S.; Petersson, E. J. J. Am. Chem. Soc. 2013, 135, 18651. doi: 10.1021/ja409709x
(13)Doose, S.; Neuweiler, H.; Sauer, M. ChemPhysChem 2009, 10, 1389. doi: 10.1002/cphc.v10:9/10
(14)Chen, H.; Ahsan, S. S.; Santiago-Berrios, M. E. B.; Abruna, H. D.; Webb, W. W. J. Am. Chem. Soc. 2010, 132, 7244. doi: 10.1021/ja100500k
(15)Gotz, M.; Hess, S.; Beste, G.; Skerra, A.; Michel-Beyerle, M. E. Biochemistry 2002, 41, 4156. doi: 10.1021/bi015888y
(16)Buschmann, V.; Weston, K. D.; Sauer, M. Bioconjugate Chem. 2003, 14, 195. doi: 10.1021/bc025600x
(17)Marme, N.; Knemeyer, J. P.; Sauer, M.; Wolfrum, J. Bioconjugate Chem. 2003, 14, 1133. doi: 10.1021/bc0341324
(18)Doose, S.; Neuweiler, H.; Sauer, M. ChemPhysChem 2005, 6, 2277.
(19)Luo, G. B.; Andricioaei, I.; Xie, X. S.; Karplus, M. J. Phys. Chem. B 2006, 110, 9363. doi: 10.1021/jp057497p
(20)Mataga, N.; Chosrowjan, H.; Shibata, Y.; Tanaka, F. J. Phys. Chem. B 1998, 102, 7081.
(21)Mataga, N.; Chosrowjan, H.; Shibata, Y.; Tanaka, F.; Nishina, Y.; Shiga, K. J. Phys. Chem. B 2000, 104, 10667. doi: 10.1021/jp002145y
(22)Mataga, N.; Chosrowjan, H.; Taniguchi, S.; Tanaka, F.; Kido, N.; Kitamura, M. J. Phys. Chem. B 2002, 106, 8917.
(23)Sun, Q. F.; Lu, R.; Yu, A. C. J. Phys. Chem. B 2012, 116, 660. doi: 10.1021/jp2100304
(24)Vaiana, A. C.; Neuweiler, H.; Schulz, A.; Wolfrum, J.; Sauer, M.; Smith, J. C. J. Am. Chem. Soc. 2003, 125, 14564. doi: 10.1021/ja036082j
(25)Zhong, D. P.; Zewail, A. H. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 11867. doi: 10.1073/pnas.211440398
(26)Zhu, R. X.; Li, X.; Zhao, X. S.; Yu, A. C. J. Phys. Chem. B 2011, 115, 5001. doi: 10.1021/jp200876d
(27)Visser, A.; van den Berg, P. A. W.; Visser, N. V.; van Hoek, A.; van den Burg, H. A.; Parsonage, D.; Claiborne, A. J. Phys. Chem. B 1998, 102, 10431. doi: 10.1021/jp982141h
(28)Laan, W.; Gauden, M.; Yeremenko, S.; van Grondelle, R.; Kennis, J. T. M.; Hellingwerf, K. J. Biochemistry 2006, 45, 51. doi: 10.1021/bi051367p
(29)Sjodin, M.; Ghanem, R.; Polivka, T.; Pan, J.; Styring, S.; Sun, L. C.; Sundstrom, V.; Hammarstrom, L. Phys. Chem. Chem. Phys. 2004, 6, 4851. doi: 10.1039/b407383e
(30)Mathes, T.; Zhu, J. Y.; van Stokkum, I. H. M.; Groot, M. L.; Hegemann, P.; Kennis, J. T. M. J. Phys. Chem. Lett. 2012, 3, 203. doi: 10.1021/jz201579y
(31)Marcus, R. A.; Sutin, N. Biochim. Biophys. Acta 1985, 811, 265. doi: 10.1016/0304-4173(85)90014-X
(32)Weinberg, D. R.; Gagliardi, C. J.; Hull, J. F.; Murphy, C. F.; Kent, C. A.; Westlake, B. C.; Paul, A.; Ess, D. H.; McCafferty, D. G.; Meyer, T. J. Chem. Rev. 2012, 112, 4016. doi: 10.1021/cr200177j
(33)Hammes-Schiffer, S.; Stuchebrukhov, A. A. Chem. Rev. 2010, 110, 6939. doi: 10.1021/cr1001436
(34)Zhang, Y.; Yuan, S. W.; Lu, R.; Yu, A. C. J. Phys. Chem. B 2013, 117, 7308.
(35)Arbeloa, E. M.; Porcal, G. V.; Bertolotti, S. G.; Previtali, C. M. J. Photochem. Photobiol. A: Chem. 2013, 252, 31. doi: 10.1016/j.jphotochem.2012.11.003
(36)Yuan, S. W.; Lu, R.; Yu, A. C. Acta Phys. -Chim. Sin. 2014, 30, 987. [袁树威, 吕 荣, 于安池. 物理化学学报, 2014, 30, 987.] doi: 10.3866/PKU.WHXB201403112
(37)Zhong, R. B.; Lu, R.; Yu, A. C. Sci. China Chem. 2013, 56, 230. doi: 10.1007/s11426-012-4788-2
(38)Lakowicz, J. R. Principles of Fluorescence Spectroscopy;Plenum Press, New York, 1999.
(39)Fita, P.; Fedoseeva, M.; Vauthey, E. J. Phys. Chem. A 2011, 115, 2465.
(40)Fiebig, T.; Wan, C. Z.; Zewail, A. H. ChemPhysChem 2002, 3, 781. doi: 10.1002/1439-7641(20020916)3:9<781::AIDCPHC781>3.0.CO;2-U
(41)Rachofsky, E. L.; Osman, R.; Ross, J. B. A. Biochemistry 2001, 40, 946. doi: 10.1021/bi001664o
(42)Hazra, A.; Soudackov, A. V.; Hammes-Schiffer, S. J. Phys. Chem. Lett. 2011, 2, 36. doi: 10.1021/jz101532g
(43)Hammes-Schiffer, S. Energy Environ. Sci. 2012, 5, 7696. doi: 10.1039/c2ee03361e
(44)Mayer, J. M. J. Phys. Chem. Lett. 2011, 2, 1481. doi: 10.1021/jz200021y
(45)Schrauben, J. N.; Cattaneo, M.; Day, T. C.; Tenderholt, A. L.; Mayer, J. M. J. Am. Chem. Soc. 2012, 134, 16635. doi: 10.1021/ja305668h
(46)Weller, A. Z. Phys. Chem. 1982, 133, 93. doi: 10.1524/zpch. 1982.133.1.093
(47)Zhang, J. B.; Sun, L. N.; Ichinose, K.; Funabiki, K.; Yoshida, T. Phys. Chem. Chem. Phys. 2010, 12, 10494. doi: 10.1039/c002831b
(48)Irebo, T.; Zhang, M. T.; Markle, T. F.; Scott, A. M.; Hammarstrom, L. J. Am. Chem. Soc. 2012, 13, 16247.
(49)Seidel, C. A. M.; Schulz, A.; Sauer, M. H. M. J. Phys. Chem. 1996, 100, 5541. doi: 10.1021/jp951507c
(50)Krichevsky, O.; Bonnet, G. Report Prog. Phys. 2002, 65, 251. doi: 10.1088/0034-4885/65/2/203
Fluorescence Quenching of Eosin Y by Tyrosine
WANG Jing-Dong LI Shuang LÜ Rong YU An-Chi*
(Department of Chemistry, Renmin University of China, Beijing 100872, P. R. China)
Quenching of a fluorescent probe by amino acid residues can provide valuable information about the structural and conformational dynamics of a biopolymer. Herein, we systematically investigated the ultrafast fluorescence quenching dynamics of Eosin Y in the presence of N-acetyl-tyrosine (AcTyr) in H2O and D2O solutions using both femtosecond transient absorption and time-correlated single-photon counting experiments. We found that the quenching of the fluorescence of Eosin Y by AcTyr in aqueous solution is mainly because of the formation of a ground-state complex between Eosin Y and AcTyr. We also found that the lifetime of the ground-state complex formed between Eosin Y and AcTyr showed a clear kinetic isotope effect, indicating that the quenching of the fluorescence of Eosin Y by AcTyr in aqueous solution is via a proton-coupled electron transfer process.
Femtosecond transient absorption spectroscopy; Time-correlated single-photon counting; Ground-state complex; Tyrosine quenching; Proton-coupled electron transfer
O643
10.3866/PKU.WHXB201507241
Received: April 17, 2015; Revised: July 24, 2015; Published on Web: July 24, 2015.
*Corresponding author. Eamil: a.yu@chem.ruc.edu.cn; Tel: +86-10-62514601; Fax: +86-10-62516444. The project was supported by the National Natural Science Foundation of China (21373269).
国家自然科学基金(21373269)资助项目
© Editorial office of Acta Physico-Chimica Sinica
