黄山大气气溶胶新粒子生长特性观测分析
2015-11-17高晋徽南京信息工程大学中国气象局气溶胶与云降水重点开放实验室江苏南京210044
郝 囝,银 燕,肖 辉,袁 亮,高晋徽,陈 魁 (南京信息工程大学,中国气象局气溶胶与云降水重点开放实验室,江苏 南京 210044)
黄山大气气溶胶新粒子生长特性观测分析
郝 囝,银 燕*,肖 辉,袁 亮,高晋徽,陈 魁 (南京信息工程大学,中国气象局气溶胶与云降水重点开放实验室,江苏 南京 210044)
利用2012年9月22日~10月28日黄山地区大气气溶胶、二氧化硫和臭氧观测数据,结合气象数据,分析气溶胶新粒子的生成-增长特征.分析发现,在33个有效观测日中,有新粒子生成-增长的观测日占总数的18.2%,其中晴天的发生频率为37.5%,新粒子生成-增长都开始于晴天上午,与无新粒子观测日相比,太阳辐射量、风速、SO2及O3浓度较高,环境温度和相对湿度较低.气溶胶新粒子的增长具有由小及大的特点,核模态气溶胶粒子(10~20nm)数浓度最先增加,爱根核模态粒子(20~50nm)数浓度随着时间推移逐渐增大,但浓度峰值依次下降,平均增长率为3.58nm/h. SO2浓度先于核模态气溶胶数浓度到达峰值,其氧化后的产物H2SO4为新粒子的核化提供前体物,并且参与新粒子的增长过程,当SO2浓度较低时,不会发生新粒子生长事件.
新粒子生长;粒子增长率;痕量气体;黄山
新粒子生长过程是影响气溶胶特性的重要因素,也是气溶胶的主要来源之一,有时与一次排放的气溶胶颗粒浓度相当[1].新粒子生成过程是指大气中低挥发性的物质达到饱和状态时冷却成核的现象,通常大气成核过程就是大气中低挥发性分子簇在气相中自然生成的过程[2].到目前为止,研究认为新粒子生成的气-粒转化机制有4种形态,二元成核[3]、三元成核[4]、离子诱导成核[5]以及有机物参与成核[6].Guo等[7]指出,在粒子的增长过程中也有化学成分的参与,包括SO2、O3、硫酸气体以及有机化合物.
近年来国内外对新粒子生成和增长进行了大量观测研究,包括在清洁大陆地区[8-10]、污染大陆地区[11-12]、沿海地区[13]以及边界层[14-16].我国对此项研究开展的较晚,主要集中在北京[17-20]、长江三角洲[21-22]、珠江三角洲[23]以及香港地区[24-25].Kulmala等[26]总结了以往的研究,在边界层内,粒径为3nm的气溶胶粒子成核率在0.01~10/(cm3·s),一般情况下城市地区的成核率要高于这个范围,基本到达100/(cm3·s),在沿海地区和工业烟羽中观测得到的成核率为104~105/(cm3·s).在中纬度地区,典型的气溶胶粒子增长率为1~20nm/h,而在极地地区,增长率只有0.1nm/h.我国观测得到的气溶胶粒子增长率在北京地区[17-20]为0.1~11.2nm/h,成核率为1.1~81.4/(cm3·s),长江三角洲地区[21-22]的粒子增长率为4.8~7nm/h,珠江三角洲地区[23]粒子增长率为2.2~19.8nm/h,粒子成核率为0.5~5.2/(cm3·s),而在香港地区[25]的粒子增长率为1.5~8.4nm/h.总体来说,污染地区较清洁地区的粒子增长率偏高.
我国关于新粒子生长的研究还需要大量的观测研究来充实.黄山地处中国东部地区,通过已有的观测研究[2,27-28]证明,黄山地区的气溶胶浓度较低,大气较为清洁,受到的污染比较轻微,大气环境属于背景大气的类型,污染物来源主要受到山谷风影响[27],这种影响大于人类活动和大气结构的影响.由于背景气溶胶浓度较低,气态污染物在液化成核后不会因背景气溶胶过多而被大量碰并消除,继而可以增长至可观测范围,有利于揭示大气气溶胶新粒子的生成及增长特征.本文采用外场观测的方法,研究黄山地区气溶胶新粒子生长特征,旨在充实我国新粒子生长的观测研究.黄山地区的研究可以补充华东背景地区新粒子生长观测研究的空白,也反映出与我国城市污染地区新粒子生长特征的鲜明对比.
1 资料与方法
1.1 观测地点、仪器介绍及数据处理
黄山(30°01′~30°18′N,118°01′~118°17′E)处在中国东部省份安徽省的南部,南北长约40km,东西宽约30km,有丰富的植被覆盖.黄山地区具有亚热带气候特征,一年四季具有春湿、夏凉、秋燥、冬寒的特点.本研究于2012年9~10月在黄山进行观测,秋季凉爽干燥,晴朗无云的天气偏多,很少有降水及云海发生.观测地点位于安徽省黄山云谷山庄(30.12°N,118.18°E),海拔高度为869m,坐落在黄山的东南方,受人类活动影响较小.
本观测使用MSP公司生产的宽粒径颗粒谱仪WPS(Wide-Range Particle Spectrometer),粒径测量范围为0.01~10μm,进行了为期37d的连续观测,观测时间为2012年9月22日~10月28日.WPS对气溶胶粒子进行分档统计,设置其时间分辨率为5min,并且使用干燥管对进入采样器的气溶胶粒子进行干燥,将气溶胶的相对湿度控制在40%以下.同时有自动气象站对温、压、湿、风及降水进行同步观测,时间分辨率设置为1min.为探究污染气体对新粒子生成的作用,还在同一时间段进行了对污染气体SO2、O3的观测.为保证数据可信可用,将WPS开机后30min以及仪器故障时的数据剔除,共得到8806组样本,在进行新粒子特征分析时,将一天中连续观测时间少于12h的观测日剔除,共得到33个有效观测日.同样将气象资料及污染气体资料也进行数据的质量控制,剔除由于开关机及仪器故障时造成的浮动较大的数据.
1.2 新粒子生成事件的判定标准
根据Maso等[10]在北欧森林连续8年的观测结果,本文采用其给出的判定新粒子生成的标准:颗粒物的数谱分布中有一个新的模态出现;这个新模态必须从核模态粒径范围开始出现;新的模态在大气中存在一段时间(几个小时);新的模态呈现增长的趋势.根据这一标准,有效观测日33d中共有6d符合这一标准,定义为新粒子日(event day),分别为9月30日,10月11日,10月12日,10月17日,10月18日和10月23日;另外,有3d定义为不明确日 (undefined day),即只满足新粒子生成标准中的部分条件,但不可定义为有新粒子生成;剩下的24d是无新粒子日(non-event day),没有新粒子事件的发生,气溶胶数浓度保持在较低的数值.
1.3 气溶胶粒子增长率的计算
气溶胶粒子的粒径增长率可以体现新粒子在增长过程中,由较小粒径段长大到较大粒径段的快慢程度.根据公式GR=ΔDm/Δt 来计算新粒子的平均增长速率,其中△Dm为新粒子增长过程中增大的粒径,△t为新粒子增长过程所经历的时间[29],不同粒径气溶胶的增长速率也具有差别,GR代表新粒子在整个增长过程的平均增长速率.
2 结果与讨论
2.1 新粒子事件的统计

表1 观测期间气溶胶粒子增长过程个例统计Table 1 The statistics of aerosol particle growth during observation
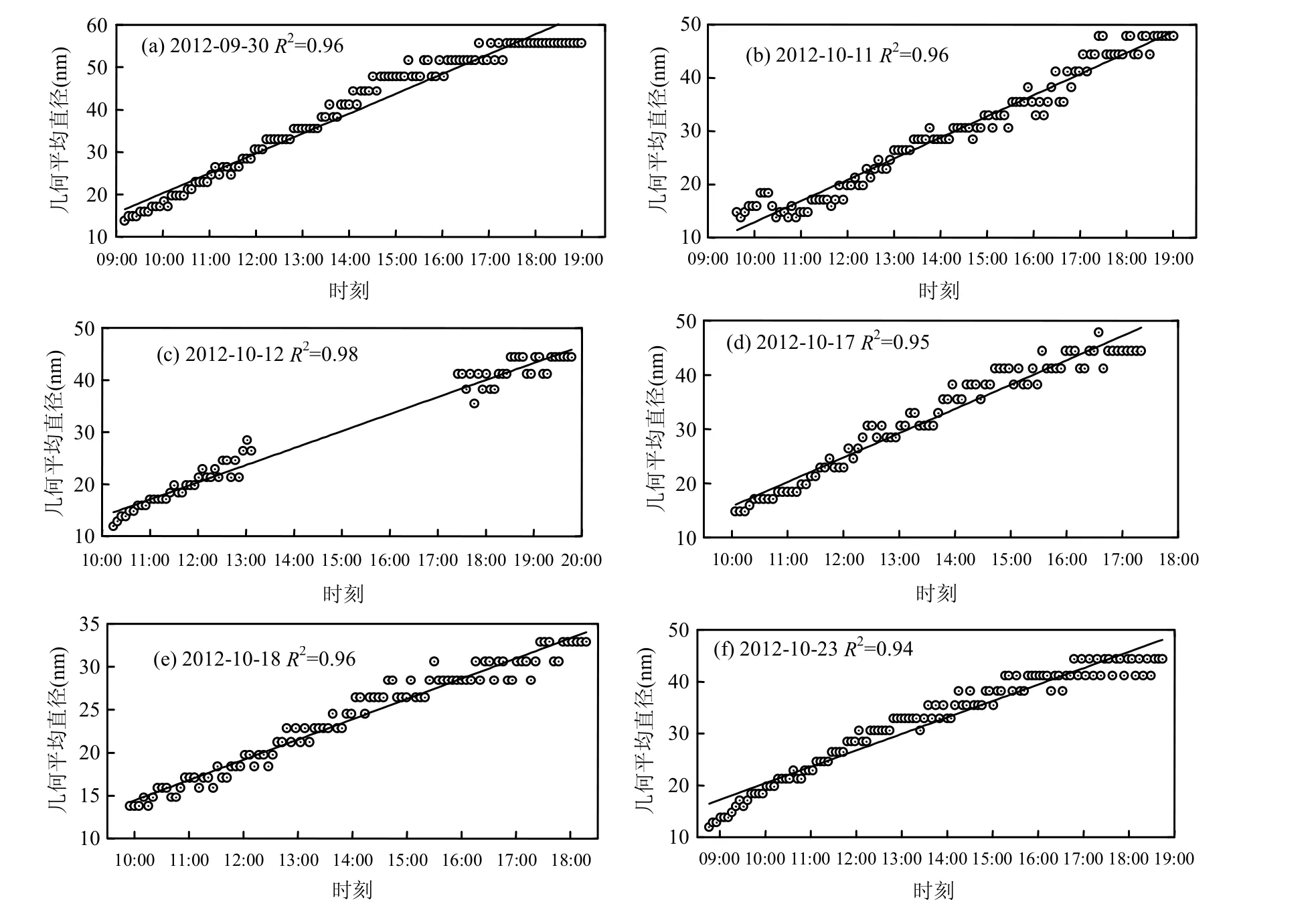
图1 颗粒物几何平均直径的变化趋势Fig.1 Tendency of the geometric mean diameter of aerosol particle
表1给出了观测期间6次新粒子增长过程的统计情况,根据新粒子增长的特征,规定开始时间为气溶胶粒子数浓度显著升高、增长量达到几个量级的时刻,结束时间为气溶胶粒子数浓度开始减少的时刻.统计发现开始时间最早为08:45,最晚为10:53,都处于太阳辐射开始增强的阶段.结束时间最早是17:20,最晚的是19:47,此时的太阳辐射非常弱,这与新粒子生成-增长需要较强的太阳辐射非常吻合[30].新粒子的增长时间长达7~10h,平均持续8.8h.根据Hamed等[12]提出的标准对此次观测进行分型发现,观测期间发生的新粒子事件都属于class I类型事件,即气溶胶粒子在增长过程中,小粒子(10~30nm)呈现出紧凑鲜明的增长特征,这说明黄山地区新粒子的生成-增长情况具有比较统一的特征.但是每个事件的增长率具有差异,范围在2.29~4.27nm/h之间,平均增长速率为3.58nm/h,与其他清洁地区[25]新粒子生成的增长速率相近,比污染地区[17-18]数值偏低.气溶胶颗粒增长率高的事件,对应粒子的最终粒径也较大,呈现出最终粒径与粒子增长率成正相关的情况.由图1可以发现,每一次新粒子事件粒子的几何平均直径与时间都具有良好的线性关系,相关系数都达到0.94以上.
2.2 新粒子事件的特征分析
2.2.1 新粒子事件个例分析 在6个新粒子事件中选取10月23日的事件进行个例分析.午夜12:00到早晨08:00之间,粒径小于50nm的气溶胶粒子数浓度保持在100cm-3以下(图2b),说明在发生新粒子生成事件之前大气中的超细粒子含量很低. 8:45 10~20nm小粒子的数浓度(N10~20)开始发生突增,在短短1h内达到峰值,数值增加了接近十倍,新粒子事件开始发生,在气溶胶数浓度随时间-粒径分布图上可见“倒香蕉”的形态(图2a),图2a中黑点所代表颗粒物的几何平均直径也表现出气溶胶粒子的增长特征.09:10 20~30nm粒径段粒子数浓度(N20~30)开始突增,在一个多小时内增长量突破2个量级.10:00 30~40nm的粒子数浓度(N30~40)开始增加,3h后达到峰值,在接下来的1h N30~40维持在3000cm-3左右.12:00 40~50nm粒子数浓度(N40~50)开始缓缓增加,增长4h后达到峰值.
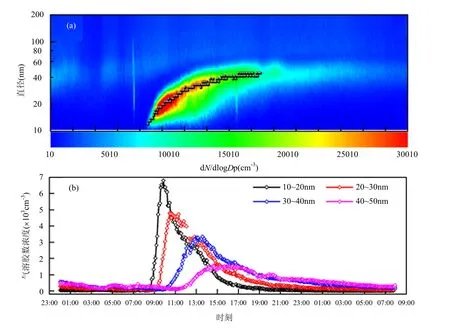
图2 (a)2012年10月23日大气气溶胶粒子的浓度分布随时间和粒径的分布及(b)不同粒径段气溶胶粒子数浓度的时间变化Fig.2 Time evolutions of the number distribution of aerosol particle (a) and 4different size ranges aerosol number concentration (b) on 23 October 2012
新粒子增长事件中气溶胶粒子的增长具有3个特点,粒子数浓度增长的开始时间随着粒子粒径增大而推迟,浓度峰值随着粒子粒径增大而减小,粒子数浓度增加的速率随粒径增大而减小.在背景洁净的大气中,过饱和蒸汽凝结为气溶胶粒子,形成小于10nm的新粒子,越来越多的新粒子通过相互碰并、凝结过饱和蒸汽而长大,出现了10~20nm粒子数浓度的增加,而更大粒子数浓度的增加都是由于10~20nm粒子的碰并、凝结作用所致.可见新粒子事件发生带来的是核模态粒子的爆发性增长,对于爱根核模态的气溶胶粒子数量的增长提供了基础.由表1、图1和图2a可以看出,在黄山生成的新粒子并没有增长到100nm以上,这与许多城市污染地区不同[17-19],说明新粒子生成现象具有局地性[12],观测地点处的背景气溶胶浓度及所受到的局地排放、远程输送的污染物浓度都会影响新粒子的生长过程.
从图3a中可以发现,太阳辐射量仅在6:00~17:00为正值,其余为0,且在09:00突然增强,在11:00~13:00维持在最高强度.太阳辐射对于新粒子的成核具有重要作用,可挥发性有机物(VOCs)会参与光化学反应生成气溶胶粒子,充足的太阳辐射为发生光化学反应提供条件. 风向在上午09:00发生转向,由偏北转为偏南,并在下午15:00发生了第2次转向,再次以偏北风作为主导.统观所有观测日,风向都具有随时间发生2次转向的特点,这体现了山地地区山谷风的特征.风速(WS)维持在1m/s以下,较小的风速有助于污染物在观测点停留较长时间,新生成的粒子也不会因此被吹散.相对湿度(RH)在全天都保持在75%以下,新粒子事件开始后下降至55%以下,这是由于在晴天RH较低的条件下有利于新粒子的生成[19,24].核模态的气溶胶数浓度(Nnuc)在新粒子事件发生之前和结束之后都保持在102cm-3量级,而爱根核模态的气溶胶数浓度(Nait)在相同时段的量级为103cm-3,新粒子事件爆发后这2个模态的气溶胶粒子数浓度剧烈增加. 这一现象说明新粒子事件是核模态粒子和爱根核模态粒子浓度突增的主要因素.
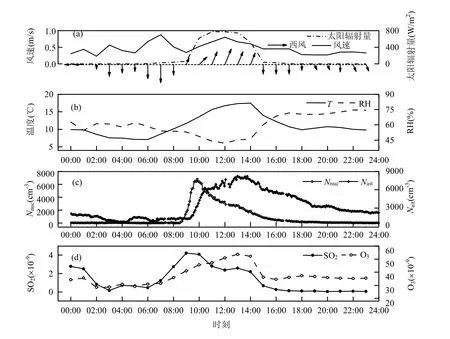
图3 2012年10月23日气象要素及污染气体的时间变化Fig.3 Hourly variations of meteorology elements and trace gases on 23 October 2012
SO2气体浓度从06:00开始增加,于09:00达到峰值,比Nnuc达到峰值的时间要提前将近1h.有研究指出新粒子的生成主要依靠SO2气体到硫化物的氧化过程,新粒子的增长也同样需要这样的过程进行凝结来维持[31].本研究观测得到的结果可以印证上述过程,SO2气体浓度达到氧化条件时,与O3发生如下化学反应:
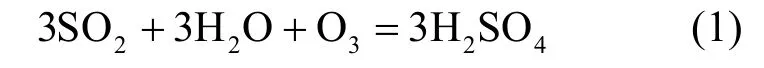
Weber 等[32]在研究海洋上空新粒子生成事件时发现,H2SO4-H2O比其他物质和水的核化现象更显著,H2SO4是超细粒子的主要前体物.本次新粒子事件也具有同样特征,可以推测H2SO4为本次新粒子生成的前体物之一.
2.2.2 气溶胶统计特征 新粒子生成事件的发生为超细粒子的数浓度提供了贡献,而无新粒子事件发生时气溶胶粒子数浓度的分布可以揭示粒子的其他来源.观测期间粒径范围在10nm~10μm的气溶胶平均数浓度为2.3× 103cm-3,与陈晨[27]、银燕等[28]和林振毅[33]对于黄山光明顶采集到的气溶胶数浓度值具有相同量级.将新粒子生成日与无新粒子生成日的气溶胶浓度进行对比(图4),发现粒子的小时分布以及谱分布具有很大的差异.在核模态(10~20nm)、爱根核模态(20~100nm)及10nm~10μm范围内的气溶胶数浓度 (Nnuc、Nait、Ntot)都在新粒子生成日偏高(图4a、b、d),积聚模态(100~1000nm)的气溶胶数浓度(Nacc)则在无新粒子生成日表现为高值(图4c).
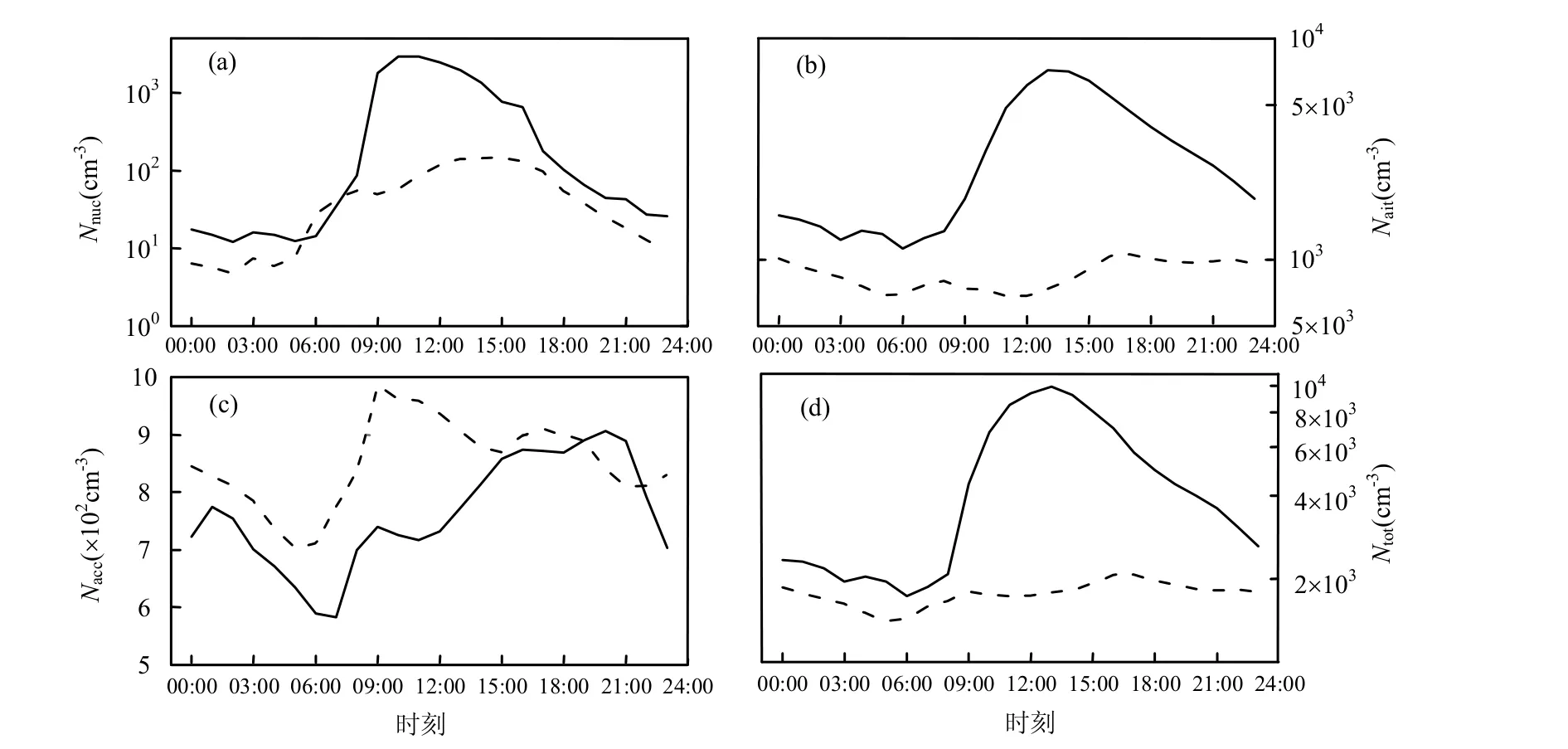
图4 新粒子日与无新粒子日不同粒径段气溶胶粒子数浓度小时平均值日变化Fig.4 Diurnal variations of hourly-averaged aerosol number concentration on event days and non-event days
核模态粒子数浓度(Nnuc) (图4a)在新粒子日从凌晨05:00开始急剧增加,从08:00开始在1h之内增加1.6×103cm-3,增长率达到最大,Nnuc在10:00~11:00达到最大值2.9×103cm-3,之后开始缓慢减小.相应的无新粒子日,Nnuc始终维持在1.5×102cm-3以下,在下午15:00达到峰值.爱根核模态粒子数浓度 (Nait)同样在新粒子日远远高于无新粒子日(图4b).Nait在新粒子日从早上06:00开始增大,10:00~11:00的增大率为各时次最大,达到1.7×103/(cm3h),Nnuc增加到13时出现最大值7×103cm-3,是Nnuc最大值的2.4倍.无新粒子日的Nait依然维持一个低值,最大值不超过1.1× 103cm-3,且出现在17:00.积聚模态气溶胶粒子数浓度(Nacc)的特征表现为在01:00~11:00在两种情况下的变化趋势基本一致,之后呈现完全不同的变化,新粒子日Nacc在11:00后维持上升的趋势,于20:00到达最大值,无新粒子日Nacc在经历先下降后上升之后在17:00到达峰值.积聚模态的气溶胶粒子主要来源于一次排放,在没有发生新粒子事件之前Nacc的变化趋势一致说明其来源一致,新粒子事件的发生致使Nacc由于小于100nm粒子的碰并增长而增加.新粒子日气溶胶粒子存在从核模态增长到爱根核模态、再增长到积聚模态的过程,无新粒子日则不存在这样的现象.
新粒子日Nait占Ntot的比例最大,对于气溶胶数浓度的贡献最大,而Nacc则是贡献最小的,无新粒子日Nait和Nacc的日平均值都有8×102cm-3,对于Ntot的贡献相当,但无新粒子日的Nacc明显高于新粒子日(图2c),这说明新粒子事件的发生需要洁净的大气背景,过多的气溶胶粒子不利于新粒子的生成和增长,一方面已经存在的气溶胶会与痕量气体发生化学反应,减少其浓度,另一方面新生成的粒子很容易与存在的气溶胶碰并消失.
2.2.3 气象要素及污染气体统计特征 新粒子生成需要在一定的气象条件下才能进行[12],图5给出了气象和气体条件在新粒子日和无新粒子日的对比.新粒子日和无新粒子日的温度(T)、相对湿度(RH)日变化趋势相同,但是新粒子日的温度和湿度在每一时刻都低于无新粒子日.新粒子日的最低温度比无新粒子日低4.3℃,最高温度低0.74℃,平均温度相差3.27℃,平均相对湿度相差14.83%.新粒子日的温度增加率和递减率都高于无新粒子日(图5a),新粒子事件发生在早晚温度差大的条件下,这与Mäkelä等[30]得到的结论相同.RH从早晨到下午的下降在新粒子日也表现得更加明显(图5b).新粒子日的太阳辐射强度明显高于无新粒子日(图5c),且在10:00~13:00维持在一个高值,这表明较强的太阳辐射有利于新粒子生成事件的发生. 不论是新粒子日还是无新粒子日,观测得到的风速都比较低(图5d),平均风速不超过0.5m/s,但新粒子日风速大于无新粒子日,尤其在早上05:00~07:00出现表现为2种不同的变化趋势.Guo等[25]在香港高山地区的观测结果表明新粒子生成时风速较大,Zhang等[34]在北京城市地区的研究认为较大的风速可以清除大气中老化的气溶胶,有助于新粒子核化后继续增长.
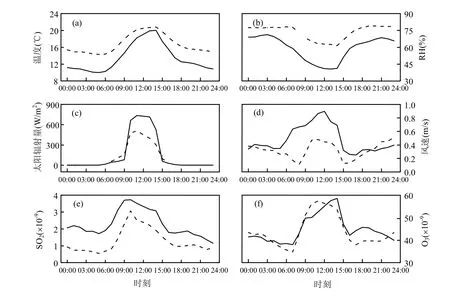
图5 新粒子日(event day)与无新粒子日(non- event day)的气象要素及污染气体浓度小时平均值日变化Fig.5 Diurnal variations of hourly-averaged meteorology elements and trace gases on event days and non-event days
SO2气体的浓度在每一时刻都表现为新粒子日高于无新粒子日,这与其他研究[12]得到的结论相同,新粒子的生成需要SO2气体浓度到达一个阈值才会发生.有无新粒子日SO2气体的浓度在01:00~09:00的变化趋势都与Nacc具有一致性(图4c,5e),SO2气体与积聚模态的气溶胶粒子来源具有一致性.新粒子日SO2气体浓度在09:00达到峰值后保持基本不变,10:00之后减小,不同的是无新粒子日SO2气体浓度在10:00才达到峰值.说明在新粒子日09:00~10:00之间SO2的浓度消耗量大于增加量,而无新粒子日不存在这样的现象.新粒子日SO2气体浓度消耗在大气成核过程中,通过参加公式(1)的化学反应生成硫酸蒸汽,可以进一步推断硫酸蒸汽为本文新粒子生成过程的前体物之一,Mäkelä 等[30]认为粒子的来源也可能与有机物活动有关. O3气体在新粒子日的平均浓度为44.92×10-9,无新粒子日为43.97×10-9,依然是新粒子日略高于无新粒子日.09:00~10:00O3气体在新粒子日的增加率很小(图5f),这一现象的解释与SO2气体情况相同.
2.3 影响新粒子事件发生的要素分析
在33个有效观测日中共有16d为晴天,12d为阴天,剩下的观测日出现了降水或雾.在16个晴天中有6d为新粒子日,2d为未定义日,剩下8d为无新粒子日,在晴天的条件下,新粒子事件的发生率为37.5%. 如果将新粒子日和未定义日视为污染日,无新粒子日视为清洁日,那么在晴天条件下,污染日和清洁日发生频率的比值为1.可见,新粒子事件全部发生在晴天,但并不是所有的晴天都有新粒子事件发生,晴天对于新粒子事件是必要而非充分条件. 不同气象要素及污染气体浓度会影响新粒子事件的发生与否,具有重要意义.
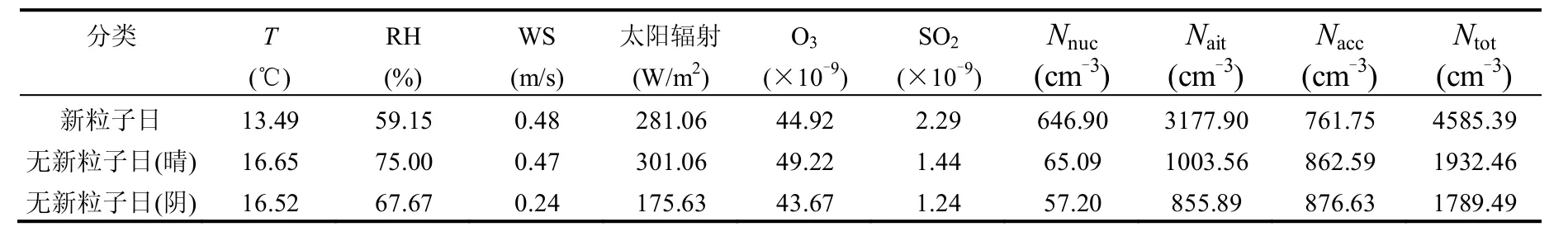
表2 新粒子日、无新粒子日(晴)和无新粒子日(阴)气象要素、污染气体、气溶胶数浓度小时平均值Table 2 Hourly averages of meteorology elementes, trace gases and aerosol concentrations on event days, sunny non-event days and cloudy non-event days
新粒子事件发生时,气象要素具有低温、低湿、高风速及强太阳辐射的特点,污染气体特征表现为SO2气体和O3气体浓度偏高.为进一步研究在晴天存在新粒子事件不发生的状况,统计了3种不同情况下的气象要素、污染气体及气溶胶粒子数浓度(表2). 通过对比气象要素发现,同样在晴天,新粒子日的气温、相对湿度和太阳辐射量低于无新粒子日,风速与无新粒子日相差不大,污染气体浓度表现为新粒子日的SO2浓度偏高而O3浓度偏低.气温、相对湿度和SO2浓度的结果与本文2.2.3结果一致,但是太阳辐射量、风速以及O3浓度不同于2.2.3的结论.太阳辐射量在新粒子日偏低6.6%,这一结果说明,太阳辐射强弱并不是新粒子事件发生的决定性条件,其只在新粒子发生时提供了反应条件,在晴天条件下没有发生新粒子生成事件的状况就可能面临反应前体物浓度不足的情况.通过计算笔者还发现,O3浓度在新粒子日比无新粒子日的晴天低8.7%,同时O3浓度在无新粒子日的阴天也偏低,且O3浓度与太阳辐射强度具有良好的正相关关系,这一结果表明即使O3浓度较高,如果反应前体物浓度偏低,新粒子核化过程还是无法发生.SO2浓度在新粒子日比无新粒子日的晴天高37%,在无新粒子日的晴天和阴天则表现为基本持平,由2.2.1可知SO2气体氧化后可以生成硫酸,硫酸是生成新粒子的重要前体物,可以推断在晴天没有发生新粒子生成事件与当天的SO2浓度偏低,氧化后生成的硫酸数量无法为新粒子生成提供充足前体物有直接关系. 以硫酸-水为主的二元成核机制在实际大气中并不是全部的成核原因,大气中的化学物种繁杂多样,很多研究[35-38]表明新粒子的成核还需要有机物(有机酸)的参与,且有机酸参与气溶胶成核是非常重要的过程[35],它会加强硫酸的核化.所以本研究中新粒子生成也存在有机物参与的可能性,晴天没有新粒子生成的原因可能包括有机物浓度不足,这一猜测还有待进一步验证. 无新粒子日的气溶胶数浓度都处于较低水平(表2),属于清洁大气状态,所以排除背景气溶胶浓度过高,核化产生的新粒子被已存在气溶胶碰并消除这一原因.
3 结论
3.1 2012年9~10月在黄山地区进行了连续37d的野外观测试验,在33个有效观测日中有6d发生了新粒子生成事件,发生时间为太阳辐射增强的时候,持续时间为7~10h,粒子的增长率为2.29~4.27nm/h,比我国城市污染地区的粒子增长率偏低60%.每一次新粒子事件粒子的几何平均直径与时间都具有良好的线性关系,R2达到0.94以上.
3.2 新粒子生成-增长事件发生时气溶胶粒子的增长具有3个特点,粒子数浓度增长的开始时间随着粒子粒径增大而推迟,浓度峰值随着粒子粒径增大而减小,粒子数浓度增加的速率随粒径增大而减小. 新粒子日10nm~10μm气溶胶数浓度平均为4585cm-3,是无新粒子日的两倍有余,比我国污染城市地区偏低一个数量级,爱根核模态粒子作为主要贡献,占据总数浓度的69%.发生新粒子事件所需的气象条件为晴朗的天气,太阳辐射较强,温度和相对湿度较低且早晚起伏大,风速在新粒子事件发生前较高,SO2气体和O3气体浓度偏高,我国城市污染地区同样反映出相同的状况.
3.3 新粒子事件在晴天条件下的发生概率为37.5%,晴天对于新粒子事件的发生是必要而非充分条件.新粒子日的太阳辐射量及O3浓度低于无新粒子日的晴天,但是SO2气体浓度比无新粒子日的晴天高37%,当SO2气体浓度偏低时,即使在晴天也不会发生新粒子事件,SO2气体浓度充足是新粒子事件发生的充要条件.
[1]Zhang R, Khalizov A, Wang L, et al. Nucleation and growth of nanoparticles in the atmosphere [J]. Chemical Reviews, 2011,112(3):1957-2011.
[2]谭 稳:华东高海拔地区大气气溶胶粒径分布及增长特征研究[D]. 南京:南京信息工程大学, 2012.
[3]Vehkamäki H, Kulmala M, Napari I, et al. An improved parameterization for sulfuric acid-water nucleation rates for tropospheric and stratospheric conditions [J]. Journal of Geophysical Research: Atmospheres (1984-2012), 2002,107(D22):AAC 3-1-AAC 3-10.
[4]Smith J N, Dunn M J, VanReken T M, et al. Chemical composition of atmospheric nanoparticles formed from nucleation in Tecamac, Mexico: Evidence for an important role for organic species in nanoparticle growth [J]. Geophysical Research Letters,2008,35(4).
[5]Kanakidou M, Seinfeld J H, Pandis S N, et al. Organic aerosol and global climate modelling: a review [J]. Atmospheric Chemistry and Physics, 2005,5(4):1053-1123.
[6]Enghoff M B, Pedersen J O P, Uggerhøj U I, et al. Aerosol nucleation induced by a high energy particle beam [J]. Geophysical Research Letters, 2011,38(9).
[7]Guo S, Hu M, Guo Q, et al. Primary Sources and Secondary Formation of Organic Aerosols in Beijing, China [J]. Environmental science and technology, 2012,46(18):9846-9853.
[8]Kristensson A, Dal Maso M, Swietlicki E, et al. Characterization of new particle formation events at a background site in Southern Sweden: relation to air mass history [J]. Tellus B, 2008,60(3):330-344.
[9]Yli-Juuti T, Nieminen T, Hirsikko A, et al. Growth rates of nucleation mode particles in Hyytiälä during 2003- 2009: variation with particle size, season, data analysis method and ambient conditions [J]. Atmospheric Chemistry and Physics,2011,11(24):12865-12886.
[10]Maso M D, Kulmala M, Riipinen I, et al. Formation and growth of fresh atmospheric aerosols: eight years of aerosol size distribution data from SMEAR II, Hyytiälä, Finland [J]. Boreal Environment Research, 2005,10(5).
[11]Dunn M J, Jiménez J L, Baumgardner D, et al. Measurements of Mexico City nanoparticle size distributions: Observations of new particle formation and growth [J]. Geophysical Research Letters,2004,31(10).
[12]Hamed A, Joutsensaari J, Mikkonen S, et al. Nucleation and growth of new particles in Po Valley, Italy [J]. Atmospheric Chemistry and Physics, 2007,7(2):355-376.
[13]O'Dowd C D, Hämeri K, Mäkelä J, et al. Coastal new particle formation: Environmental conditions and aerosolphysicochemical characteristics during nucleation bursts [J]. Journal of geophysical research, 2002,107(D19):8107.
[14]Birmili W, Wiedensohler A. New particle formation in the continental boundary layer: Meteorological and gas phase parameter influence [J]. Geophysical Research Letters, 2000,27(20):3325-3328.
[15]Nilsson E D, Paatero J, Boy M. Effects of air masses and synoptic weather on aerosol formation in the continental boundary layer [J]. Tellus B, 2001,53(4):462-478.
[16]Dal Maso M, Kulmala M, Lehtinen K E J, et al. Condensation and coagulation sinks and formation of nucleation mode particles in coastal and boreal forest boundary layers [J]. Journal of Geophysical Research: Atmospheres (1984-2012), 2002,107(D19):PAR 2-1-PAR 2-10.
[17]Yue D, Hu M, Wu Z, et al. Characteristics of aerosol size distributions and new particle formation in the summer in Beijing[J]. Journal of Geophysical Research: Atmospheres (1984-2012),2009,114(D2).
[18]Yue D L, Hu M, Zhang R Y, et al. The roles of sulfuric acid in new particle formation and growth in the mega-city of Beijing [J]. Atmospheric Chemistry and Physics, 2010,10(10):4953-4960.
[19]Wu Z, Hu M, Liu S, et al. New particle formation in Beijing,China: Statistical analysis of a 1-year data set [J]. Journal of Geophysical Research: Atmospheres (1984-2012), 2007,112(D9).
[20]Shen X J, Sun J Y, Zhang Y M, et al. First long-term study of particle number size distributions and new particle formation events of regional aerosol in the North China Plain [J]. Atmospheric Chemistry and Physics, 2011,11(4):1565-1580.
[21]Gao J, Wang T, Zhou X, et al. Measurement of aerosol number size distributions in the Yangtze River delta in China: Formation and growth of particles under polluted conditions [J]. Atmospheric Environment, 2009,43(4):829-836.
[22]Herrmann E, Ding A J, Petäjä T, et al. New particle formation in the western Yangtze River Delta: first data from SORPES-station[J]. Atmospheric Chemistry and Physics Discussions, 2013,13(1): 1455-1488.
[23]Liu S, Hu M, Wu Z, et al. Aerosol number size distribution and new particle formation at a rural/coastal site in Pearl River Delta(PRD) of China [J]. Atmospheric Environment, 2008,42(25): 6275-6283.
[24]Yao X, Choi M Y, Lau N T, et al. Growth and shrinkage of new particles in the atmosphere in Hong Kong [J]. Aerosol Science and Technology, 2010,44(8):639-650.
[25]Guo H, Wang D W, Cheung K, et al. Observation of aerosol size distribution and new particle formation at a mountain site in subtropical Hong Kong [J]. Atmospheric Chemistry and Physics,2012,12(20):9923-9939.
[26]Kulmala M, Vehkamäki H, Petäjä T, et al. Formation and growth rates of ultrafine atmospheric particles: a review of observations[J]. Journal of Aerosol Science, 2004,35(2):143-176.
[27]陈 晨.黄山大气气溶胶观测研究 [D]. 南京:南京信息工程大学, 2009.
[28]银 燕,陈 晨,陈 魁,等:黄山大气气溶胶微观特性的观测研究 [J]. 大气科学学报, 2010,33(2):129-136.
[29]Heintzenberg J. Properties of the log-normal particle size distribution [J]. Aerosol Science and Technology, 1994,21(1): 46-48.
[30]Gao J, Chai F, Wang T, et al. Particle number size distribution and new particle formation (NPF) in Lanzhou, Western China [J]. Particuology, 2011,9(6):611-618.
[31]Hoppel W A, Frick G M, Fitzgerald J W, et al. Marine boundary layer measurements of new particle formation and the effects nonprecipitating clouds have on aerosol size distribution [J]. Journal of Geophysical Research: Atmospheres (1984-2012),1994,99(D7):14443-14459.
[32]Weber R J, McMurry P H, Eisele F L, et al. Measurement of expected nucleation precursor species and 3-500-nm diameter particles at Mauna Loa observatory, Hawaii [J]. Journal of the atmospheric sciences, 1995,52(12):2242-2257.
[33]林振毅.黄山顶大气气溶胶及云雾微观特性观测分析 [D]. 南京:南京信息工程大学, 2010.
[34]Zhang Y M, Zhang X Y, Sun J Y, et al. Characterization of new particle and secondary aerosol formation during summertime in Beijing, China [J]. Tellus B, 2011,63(3):382-394.
[35]Zhang R, Suh I, Zhao J, et al. Atmospheric new particle formation enhanced by organic acids [J]. Science, 2004,304(5676):1487-1490.
[36]Guo H, Wang D W, Cheung K, et al. Observation of aerosol size distribution and new particle formation at a mountain site in subtropical Hong Kong [J]. Atmospheric Chemistry and Physics,2012,12(20):9923-9939.
[37]Hao L Q, Romakkaniemi S, Yli-Pirilä P, et al. Mass yields of secondary organic aerosols from the oxidation of α-pinene and real plant emissions [J]. Atmospheric Chemistry and Physics,2011,11(4):1367-1378.
[38]Metzger A, Verheggen B, Dommen J, et al. Evidence for the role of organics in aerosol particle formation under atmospheric conditions [J]. Proceedings of the National Academy of Sciences,2010,107(15):6646-6651.
Observation of new particle formation and growth on Mount Huang.
HAO Jian, YIN Yan*, XIAO Hui, YUAN Liang, GAO Jin-hui, CHEN Kui (Key Laboratory for Aerosol-Cloud-Precipitation of China Meteorological Administration, Nanjing University of Information Science and Technology, Nanjing 210044, China). China Environmental Science, 2015,35(1):13~22
Growth of newly formed particles on Mount Huang in eastern China was investigated using the measured aerosol particle data, trace gas and meteorology data over the period from 22 September to 28 October, 2012. The new particle formation (NPF) events appeared on 6 out of 33 days, occurring before the noon of sunny days. Moreover, the occurrence frequency of the NPF events during sunny days was 37.5%. Compared with other days (non-NPF days), solar radiation, wind speed, SO2and O3concentrations were higher in NPF days on average with lower temperature and relative humidity (RH). The nucleation mode particle (10~20nm in diameters) concentrations increased first, and then the Aiken nuclei mode particle concentrations (20~50nm in diameters) increased over time. However, the peaks of large aerosol particle concentration were lower. The mean growth rate (GR) of the newly formed aerosol particles was estimated as 3.58nm/h. SO2concentration reached the peak faster than nucleation mode particle number concentration does, and the oxidation of SO2produced H2SO4which was thought to participating in the particle nucleation as a kind of precursor. So the nucleation won’t appear under low concentration of SO2even on sunny days.
new particle formation;growth rate of particles;trace gas;Mount Huang
X51
A
1000-6923(2015)01-0013-10
郝 囝(1988-),女,内蒙古赤峰人,南京信息工程大学博士研究生,主要从事大气气溶胶环境气候效应研究.
2014-05-12
国家自然科学基金项目(41030962);江苏高校优势学科建设工程项目(PAPD);江苏省普通高校研究生科研创新计划项目(N0782002251)
* 责任作者, 教授, yinyan@nuist.edu.cn
