HNO3氧化改性对国产PAN基炭纤维表面成分和组织结构的影响
2015-10-13刘云启赵赟豪易茂中葛毅成
李 婵,武 帅,刘云启,吴 皇,赵赟豪,易茂中,葛毅成
HNO3氧化改性对国产PAN基炭纤维表面成分和组织结构的影响
李 婵,武 帅,刘云启,吴 皇,赵赟豪,易茂中,葛毅成
(中南大学粉末冶金国家重点实验室,长沙410083)
采用HNO3对国产PAN基炭纤维进行表面改性处理。采用氧氮氢联测仪、XPS、FTIR、Raman、SEM检测改性后炭纤维表面活性基团和微观结构的变化。结果表明:经55 ℃氧化处理后,纤维质量减小;而经80 ℃和100 ℃处理后,纤维质量增加。氧化处理后,纤维整体和表面的氧含量都增加,整体的氧含量明显低于表面,而增幅却高于表面。在氧化过程中,—OH、C—O、C=O、COOR及吡啶型氮、四价氮、—NO2的生成与转变同时发生,形成了低活性基团向高活性基团转变的动态过程,且随氧化温度升高或时间延长,纤维表面无序度降低。
国产PAN炭纤维;氧化改性;官能团;微观结构
PAN基炭纤维作为一种高性能的增强材料,在航空航天、医疗器械、土木建筑等领域得到广泛应用[1−2]。一般而言,未进行表面处理的炭纤维表面光滑、惰性大、表面能低、活性差、与基体浸润性差[3],影响纤维的增强效果。因此,在炭纤维使用前,对其进行表面改性处理,增强其表面活性、改善其表面结构成为必要的技术途径[4−5]。
目前,针对PAN基炭纤维的表面处理主要分为氧化法和非氧化法两大类。其中,表面氧化处理方法包括气相氧化、化学氧化和电化学氧化;非氧化法包括涂层法和聚合物接枝法[5]。
LEE[6]等采用空气氧化对T300炭纤维表面进行处理,发现处理后纤维表面官能团、表面积和微孔数量显著增加,表面能提高;PAMULA[4]等对III型PAN基炭纤维进行HNO3氧化处理,发现纤维表面吡啶型氮和四价氮经氧化处理后转变为脂类官能团,表面的氧含量是整体材料氧含量的1.5倍;FUKUNAGA[7]等对高模炭纤维进行等离子处理,发现处理后炭纤维表面的微晶尺寸变小、比表面积增加、表面活性单元明显增多。
我国高性能炭纤维的制备技术水平与国外有一定差距,如在航天构件上多使用进口炭纤维,严重制约我国民用工业、高新技术和国民经济的发展。近年来,随着国内相关研究、制备技术水平的提升,部分纤维的性能已达到或接近国外同类产品的技术水平[8]。因此,采用国产炭纤维替代进口纤维,确保相关行业的健康发展已成为可能并成为共识。然而目前针对国产炭纤维表面改性的探讨较少,在一定程度上影响了相关增强材料的制备和机理研究,限制了国产炭纤维的应用。所以,本实验在研究利用国产炭纤维替代进口材料生产炭纤维增强复合材料过程中,采用浓HNO3对国产PAN基炭纤维进行表面改性,探讨炭纤维在不同条件处理后表面成分和微观结构的变化,分析表面化学结构的演变机理,及其对后续复合材料性能可能的影响,以期为日后研究提供一定的参考。
1 实验
1.1 实验原料
实验原料为威海拓展纤维有限公司生产的CCF700炭纤维,其基本性能参数如下:丝束12 k,拉伸强度4.7 GPa,拉伸模量240 GPa,伸长率1.96%。硝酸为株洲星空化玻有限公司生产,分析纯。
1.2 改性处理
通常在炭纤维制备过程中,为了保护纤维表面在卷绕、织造等工艺中不受损伤,同时为提高特定复合材料的层间剪切强度,纤维表面会进行上浆处理[9]。因此,为了避免浆料涂层对本实验的影响,在对炭纤维进行表面改性研究之前,先在丙酮中进行索氏抽提48 h以上,以完全去除纤维表面的上浆剂及附着杂质。
将经过索氏抽提处理的炭纤维分为3组,分别浸泡在55、80、100 ℃的浓硝酸中进行氧化。氧化时间分别为30、60、90和120 min。氧化处理后的炭纤维用去离子水洗涤至中性,经80 ℃、120 min真空干燥处理后备用。
1.3 分析与测试
采用FA2104N型电子天平(精度0.000 1 g)测量氧化处理前后炭纤维的质量。采用TCH600氮氧氢分析仪测量炭纤维的整体氮氧含量,K-Alpha 1063型X-ray光电子能谱仪鉴别炭纤维表面化学元素,Nicolet 6700型傅立叶变换红外光谱仪(4000~600 cm−1)测试炭纤维的红外光谱。采用2abRAM Aranis型拉曼光谱仪(波长532 nm)检测炭纤维的表面结构,Nova Nano SEM230型场发射扫描电子显微镜观察纤维表面 形貌。
2 结果与讨论
2.1 质量变化
在氧化处理过程中,硝酸能够刻蚀纤维表面的炭乱层结构,造成较大炭层的断裂及纤维的质量损失;同时,硝酸还会氧化炭层边缘以及棱角处的不饱和碳原子,增加纤维表面活性官能团含量,使纤维质量增加。炭纤维表面的氧化刻蚀可增加表面的粗糙度,提高纤维与基体材料的机械结合,但过度刻蚀将降低纤维强度[6];增加纤维表面活性基团可以增加纤维与树脂间的化学结合,但过多杂乱分布的基团也会影响纤维表面结构的有序度,反而不利于增强复合材料[10]。氧化处理前后纤维质量的变化规律将由上述两个因素共同控制。
质量变化率如公式(1)所示:

式中:0—氧化处理前纤维质量;t—氧化处理后纤维质量。
表1所列为经不同温度和时间氧化处理后炭纤维的质量变化率。由表可知,经55 ℃处理后,炭纤维的氧化质量损失更明显;而随反应时间延长,其质量损失率变小,在处理90 min后基本达到反应平衡状态。但在80 ℃和100 ℃处理后,炭纤维官能团的增加大于氧化质量损失的影响,纤维质量增加且其质量增加率随处理时间的延长或温度的升高而增加。其中,在 100 ℃、120 min处理后纤维的质量增加最明显。

表1 炭纤维处理前后的质量变化率
Note:0—Mass of carbon fibrebefore oxidation;t—Mass of carbon fibre after oxidation;—Transform rate of mass of carbon fibre
2.2 化学元素分析
炭纤维表面的化学活性以其化学活性基团的浓度表示。一般认为,这些活性基团主要为C—O、C=O、O—C=O、N—H、N—O,所以可以用(=(O1s+N1s)/ C1s)值表示其化学活性。值越大,表明炭纤维的表面活性越强,其与树脂基体的化学结合越好,得到的复合材料的性能越高[11]。
图1(a)、(b)、(c)所示分别为HNO3氧化处理后炭纤维的XPS谱及其C1s和N1s峰的拟合曲线。由图可见,可以由结合能在284 eV处的C1s、535 eV的O1s和400~405 eV处的N1s峰强计算出C、O和N的含 量[12−13]。拟合曲线上不同结合能处的峰代表纤维表面不同的化学结构。其中,284.8 eV处C1s的主峰代表表面的C—C结构,286.4 eV的峰代表可能存在的羟基、醚键等结构(C—OR),287.5 eV处的峰代表羰基、醌基中的C=O,289.2 eV处的峰代表羧酸或酯基中的O=C—OR[6, 13]。对N1s峰拟合后发现纤维表面存在3种成分的氮,在398.4 eV处的峰代表吡啶型氮,400~401eV处的峰代表四阶氮或质子氮(—NH/—NH2),405.6 eV处的峰代表氧化型氮(—NO2)[9, 13−14]。
氧化改性处理前后炭纤维表面的C、O、N含量及(O1s+N1s):C1s如表2所列。从表中可以看出,当氧化时间一定时,随氧化温度升高,纤维表面碳含量降低,氧含量从未处理时的8.54%增加到16.54%,增幅接近1倍;氮含量比未处理时增加,在55 ℃、90 min处理后达到最高,值从13.98%增加到26.60%。而当纤维在80 ℃的HNO3中处理时,表面氧的含量比未处理时增加,增幅低于80%;随氧化时间延长,氧含量从 60 min处理后的14.99%下降到120 min后的13.72%。氮含量有波动,在60 min处理后略有下降,90 min后达3.87%,120 min后增到4.10%。

图1 氧化处理后炭纤维的XPS全谱及其C1s、N1s分峰拟合谱
因此,在提高氧化温度时,氧含量及值的增幅更明显。这表明:对纤维表面活性基团增加主要的影响因素是氧化温度,其次是反应时间。而当氧化温度一定时,反应超过一定时间后,表面的活性基团总量达到饱和;继续延长反应时间,官能团会发生分解和转化,活性基团减少。与含氧官能团的变化不同,纤维表面的氮含量在反应过程中增加不明显,55 ℃处理后达到最大值,表明氧化处理只会在纤维表面引入少量的含氮基团;在反应过程中,某些含氮结构会被破坏分解而使氮含量降低。
一般认为炭纤维表面存在许多微孔和缺陷[12],改性处理过程中酸溶液会进入这些缺陷中并与纤维发生反应。由于光电子能谱仪(XPS)只能探测到炭纤维表层10 nm深度范围以内的成分。因此,需要结合氧化前后纤维整体的C、O、N含量来分析氧化处理对表面的影响。
炭纤维整体的C、O、N含量如表2所列。由表可知,整体的氧含量明显低于表面。当氧化处理时间一定时,随氧化温度升高,整体氧含量从未处理时的0.14%增加到100 ℃、90 min处理后的0.49%,增幅超过200% ;氮含量从未处理时的4.34%增加到4.81%。而当炭纤维在80 ℃的HNO3中处理时,随氧化时间延长,氧含量从60 min的0.20%增加到120 min的0.33%,增幅超过60%;氮含量在60 min处理后略有下降, 90 min处理后含量最高。
对比可知,炭纤维的氧主要集中在纤维表面,氧化反应深度超过XPS的探测深度;随处理时间延长或氧化温度升高,氧化反应会沿表面的孔隙或缺陷向纤维内部发展。同时,炭纤维整体的氮含量增幅不明显,其摩尔含量略低于表面的氮含量,表明氧化处理过程中含氮基团的引入只发生在纤维表面很小的尺度范围内。
实验结果表明,炭纤维经氧化处理后,在表面引入大量活性基团,其中,部分含氧基团会在纤维表面较深范围内生成,从而可以增强纤维与树脂间的界面结合强度,提高复合材料的性能。
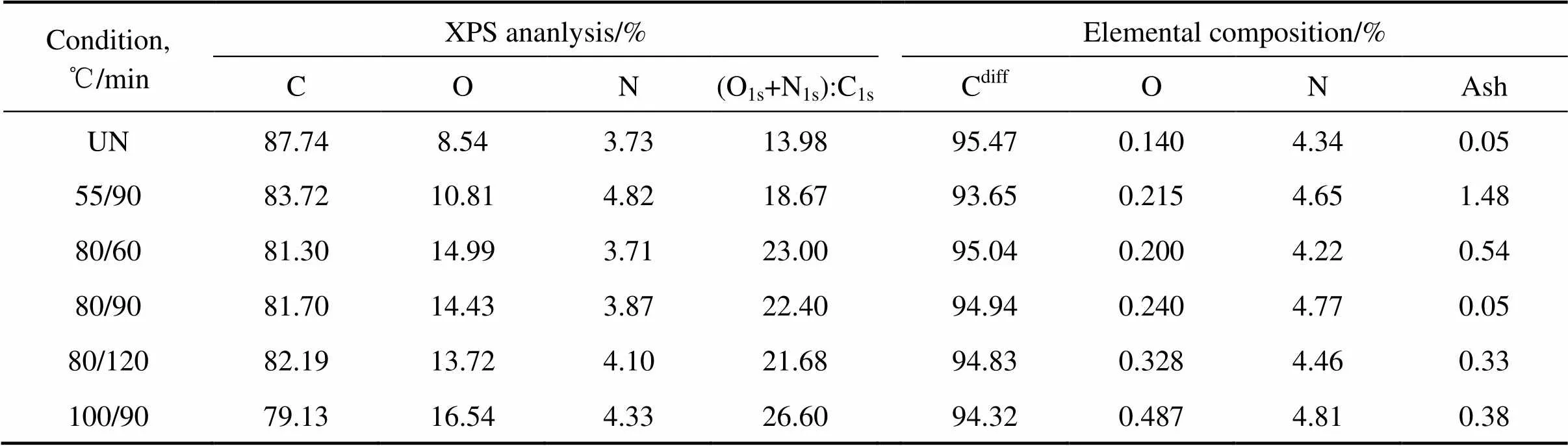
表2 氧化处理前后炭纤维的元素成分
UN—Untreated; Cdiff—obtained by difference

表3 XPS法测得氧化改性前后炭纤维的表面组成
BDL—Below detection limit; Na—Pyridine-type N; Nb—Protonated or quaternary N
2.3 化学官能团分析
C1s峰和N1s峰的拟合计算结果如表3所列。HNO3氧化处理后,炭纤维表面的C—C键结构含量明显减少。当氧化时间一定时,随氧化温度升高,C—OR和C=O的含量先增加后降低,在100 ℃、90 min处理后C=O含量略有下降;COOR的含量从未处理时的3.99%增到7.97%;吡啶型氮含量明显减少,最后消失;表面生成新的氧化型氮(—NO2),含量随温度的上升而降低。而当炭纤维在80 ℃硝酸处理时,随氧化时间延长,表面的C—O和C=O含量均先降低后增加,处理120 min后,C—O含量从11.19%降到10.41%;COOR含量从氧化60 min后的5.34%增加到氧化 120 min后的6.62%;—NH/—NH2含量先下降,处理120 min后升到3.20%;—NO2含量先增加后降低。
图2和图3分别是不同温度和不同时间氧化处理后炭纤维的红外光谱。由文献可知[12, 15],1 633 cm−1、 3 433 cm−1是羟基(—OH)的弯曲和伸缩振动峰, 1 720 cm−1是C=O键伸缩振动峰,1 150 cm−1, 1 068 cm−1是酒精、酚和醚中的C—O键振动峰, 1 380 cm−1是羧基的C—O键的振动峰。从图中可以看出,炭纤维在氧化时间一定时,随氧化温度升高,—OH的特征峰先减弱后增强,100 ℃时明显降低;C—O明显减少,1 380 cm−1处C—O单键略有增加;C=O振动峰基本消失。在氧化温度一定时,随氧化时间延长,—OH的特征峰先减弱后增强;C—O的特征峰强度先减弱,90 min时明显增强,120 min时又显著下降;1 380 cm−1处C—O单键略有增加;C=O键振动峰基本消失。

图2 不同温度氧化炭纤维的FTIR光谱图

图3 氧化不同时间炭纤维的FTIR图
综上可知,在氧化初始阶段,纤维表面的—OH被氧化为C—O或C=O,造成FTIR中—OH特征峰减弱;同时,纤维表面活性碳原子被氧化生成新的—OH,使—OH的特征峰增强。C—O的含量变化有波动,整体上有减少的趋势。说明氧化过程中C—O被氧化为C=O或COOR;C=O的含量除了在55 ℃处理时有升高,其它条件下都下降。这说明在氧化过程中C=O被氧化为COOR,且COOR的含量随氧化温度升高或时间延长而增加。这表明,炭纤维在处理过程中,表面含氧基团的演变是低活性基团向更高活性基团转变的动态过程,反应过程如图4所示。因此,提高含氧基团的活性,可以增大纤维表面与树脂化学结合键的强度,从而提高复合材料的强度。
同时,随氧化温度升高或时间延长,纤维表面的吡啶型氮持续减少直至消失,并且出现了新的氧化型氮,四阶氮或质子氮的含量也略有增加。说明在硝酸氧化过程中,吡啶型氮会被转化为四阶氮或氧化型氮,且在表面直接引入氧化型氮。反应过程如图5所示。
2.4 Raman光谱分析
图6所示为硝酸氧化处理后炭纤维表面的Raman光谱。在炭纤维拉曼光谱区域内存在两条主要谱线:D线(1 360 cm−1)和G线(1 580 cm−1)。其中,D峰是由炭材料的无序结构引起;G峰可用来表征炭材料中SP2键的完整程度。此外,在1 150~1 200 cm−1范围的D″峰是脂肪结构或类烯烃结构中碳−碳键的伸缩振动引起的;1 500~1 550 cm−1范围的A峰一般认为是由无定型碳或者某些有机官能团的存在引起;石墨微晶的E2g振动引起的D′峰在拉曼谱线的1 620 cm−1附 近[16−17]。D峰强度D和G峰强度G比值(=D/G)可用来判断纤维表面炭层的有序程度。

图4 氧化处理炭纤维表面含氧基团转化示意图

图5 氧化处理炭纤维表面含氮基团转化示意图
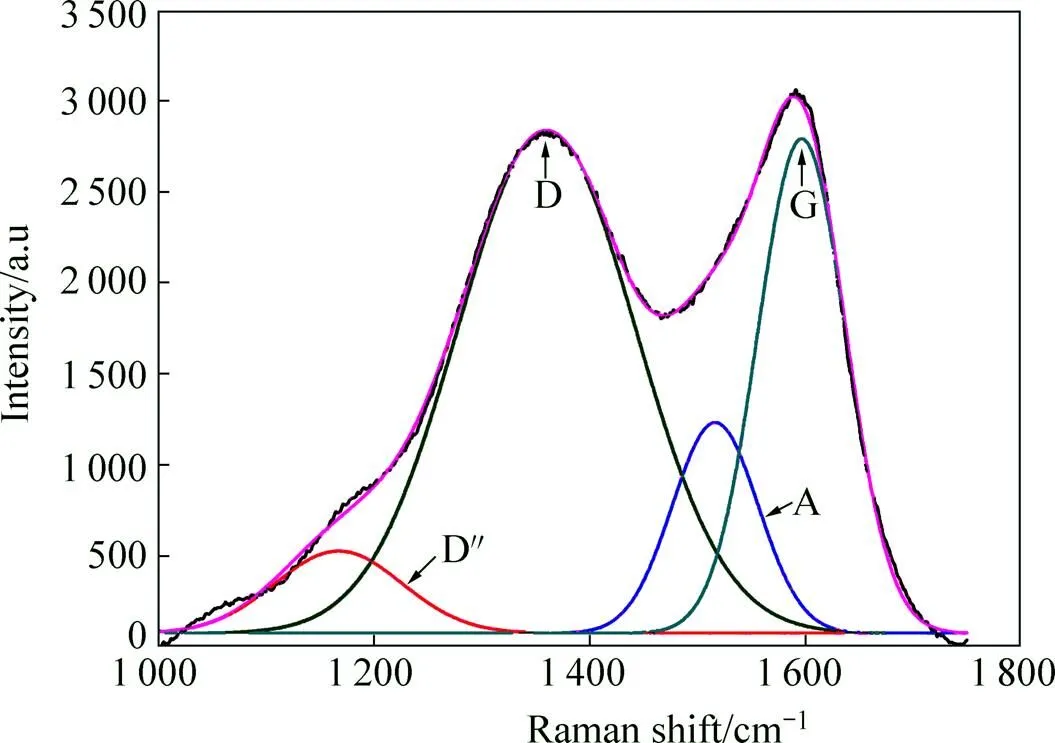
图6 硝酸氧化处理炭纤维的Raman光谱拟合曲线
由Raman光谱的拟合曲线可知,炭纤维未经高温炭化/石墨化处理时,表面完整的石墨微晶较少,D′峰难以拟合出来。Guassian拟合结果如表4所列。从表中可以看出,随氧化温度升高或时间延长,代表无序结构的D线的峰位向低峰位偏移,半高宽减小,积分强度比(=D/G)降低;D″线的峰位基本不变,半高宽显著下降,积分强度比略有下降;A线的峰位向高峰位偏移,半高宽增加,积分强度比A/G从0.16增加到0.44。
上述结果表明,未氧化处理的炭纤维的表面结构不完善,存在较多缺陷和无序碳原子。处理后,表面不稳定和无序排列的碳原子被氧化,一定程度上增加了纤维表面的有序度;在纤维表面棱角、孔隙和缺陷处生成有机官能团,修补了一部分表面缺陷,改善了纤维表面的微观结构。所以,纤维经适度氧化处理可在一定程度上提高其力学性能。
2.5 表面形貌
图7为氧化前后炭纤维的表面形貌。由图可见,未处理炭纤维表面分布较多平行轴向的浅沟槽。经 55 ℃/60 min氧化处理后,纤维的表面产生了不均匀的轻微氧化刻蚀,表面沟槽稍有加深。这表明,经氧化处理后,炭纤维表面的波纹度、比表面积和表面的粗糙度增加,可提高复合材料界面的机械结合力。经氧化处理120 min后,纤维表面部分区域发生严重氧化刻蚀,甚至剥落形成表面凹坑。这说明,过度氧化会破坏纤维表面结构的完整性,影响自身的机械强度,反而不利于增强复合材料,所以在对炭纤维改性处理时,需要选择合适的实验参数。
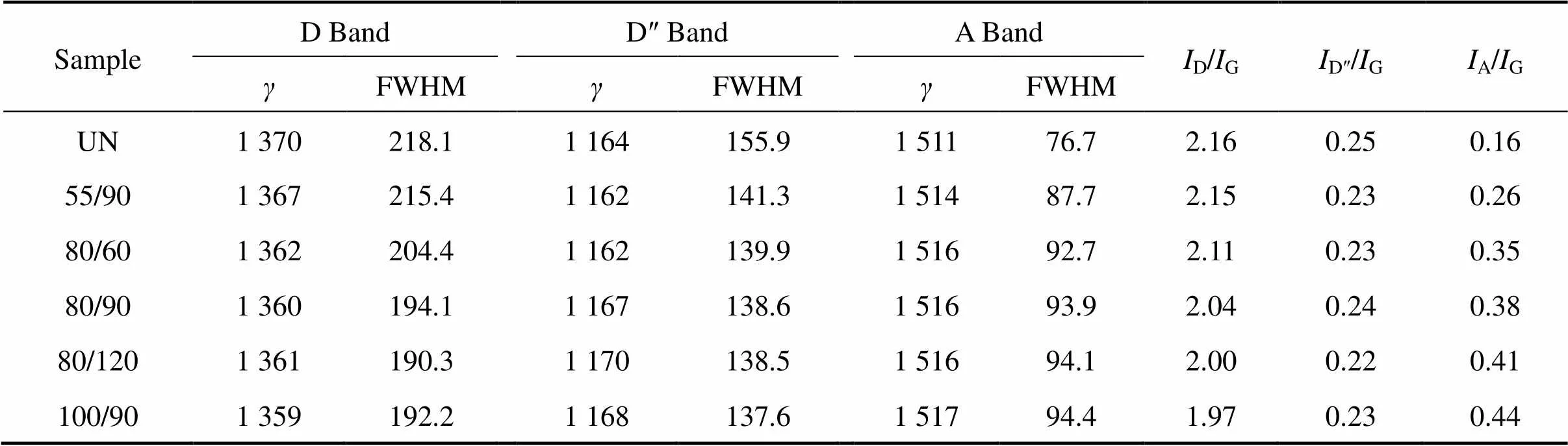
表4 表面处理炭纤维的拉曼分析

图7 氧化处理前后炭纤维的表面形貌
3 结论
1) 国产PAN基炭纤维经55 ℃氧化处理后,纤维质量减小,随反应时间延长,其质量损失率减小;但80 ℃和100 ℃处理后,纤维质量增加且质量增加率随处理时间延长或温度升高而增大。
2) 当氧化时间一定时,随温度升高,纤维整体和表面的氧含量均增加,整体的含量明显低于表面,但增幅高于表面,氮含量从未处理时的4.34%持续增加到4.81%;经80 ℃处理后,随氧化时间延长,整体氧含量增加,但表面氧含量略有下降,氮含量变化有 波动。
3) 炭纤维经氧化处理后,会在表面引入新的—OH,同时—OH会转变为C—O或C=O结构,造成C—O、C=O含量增加;C—O、C=O会向活性更高的COOR发生转化,造成C—O、C=O含量的降低,COOR含量增加,形成了低活性基团向更高活性基团转变的动态过程。同时,随氧化温度升高或时间延长,纤维表面无序度降低。
REFERENCES
[1] 上官倩芡, 蔡泖华. 碳纤维及其复合材料的发展及应用[J]. 上海师范大学学报(自然科学版), 2008, 37(3): 275−278. SHANGGUAN Qian-qian, CAI Mao-hua. Development and applications of carbon fiber and its composites [J]. Journal of Shanghai Normal University (Natural Sciences), 2008, 37(3): 275−278.
[2] 王林山, 熊 翔, 李江鸿, 等. 国内C/C复合材料的研究发展现状[J]. 粉末冶金材料科学与工程, 2001, 6(2): 118−122. WANG Lin-shan, XIONG Xiang, LI Jiang-hong, et al. The Present status of carbon-carbon composites [J]. Materials Science and Engineering of Powder Metallurgy, 2001, 6(2): 118−122.
[3] 袁 华, 王成国, 卢文博, 等. PAN基碳纤维表面液相氧化改性研究[J].航空材料学报, 2012, 32(2): 65−68. YUAN Hua, WANG Cheng-guo, LU Wen-bo. Liquid-phase oxidation modification of PAN-based carbon fiber surface [J]. Journal of Aeronautical Materials, 2012, 32(2): 65−68.
[4] ELZBIETA P, PAUL G R. Bulk and surface chemical functionalities of type III PAN-based carbon fibers [J]. Carbon, 2003, 41(10): 1905−1915.
[5] VICKERS P, WATTS J F, PERRUCHOT C, et al. The surface chemistry and acid-base properties of a PAN-based carbon fibre [J]. Carbon, 2000, 38(5): 675−89.
[6] LEE J S, KANG T J. Changes in physic-chemical and morphological properties of carbon fiber by surface treatment [J]. Carbon, 1997, 35(2): 209−216.
[7] FUKUNAGA A, KOMAMI T, UEDA S, et al. Plasma treatment of pitch-based ultra high modulus carbon fibers [J]. Carbon, 1999, 37: 1087−1091.
[8] 张凤翻. 国产碳纤维规模化生产及应用值得注意的几个问题[J]. 高科技纤维与应用, 2005, 30(6): 1−6, 13. ZHANG Feng-fan. The concerns need to be considered on the industrial applications of the domestic carbon fiber [J].Hi-Tech Fiber & Application, 2005, 30(6): 1−6, 13.
[9] 邵友林, 王伯羲. 碳纤维的表面除胶及表征[J]. 复合材料学报, 2002, 19(4): 29−32. SHAO You-lin, WANG Bo-yi. Surface Desizing and indication of carbon fiber [J]. Acta Material Composite Sinica, 2002, 19(4): 29−32.
[10] MANFRED P, GERHARD K, HERWIG P. Structural investigation of carbon/carbon composites by neutron scattering [J]. Physica, 2006: 538−541.
[11] CHUKOV D I, STEPASHKIN A A, GORSHENKOV M V, et al. Surface modification of carbon fibers and its effect on the fiber-matrix interaction of uhmwpe based composites [J]. Journal of Alloys and Composites, 2014, 586: s459−s463.
[12] ZIELKE U, HUTTINGER K J, HOFFMAN W P. Surface oxidized carbon fibers: II. chemical modification [J]. Carbon, 1996, 34(8): 999−1005.
[13] XU Bing, WANG Xiao-shu, LU Yun. Surface modification of polyacrylonitrile-based carbon fiber and its interaction with imide [J]. Applied Surface Science, 2006, 253(5): 2695−2701.
[14] LEE W H, LEE J G, PEUCROFT P G. XPS study of carbon fiber surfaces treated by thermal oxidation in a gas mixture of O2/(O2+N2) [J]. Applied Surface Science, 2001, 171: 136−142.
[15] CHLOPEK J, CHOCHOL A M, PALUSZKIEWICZ C. FTIR evaluation of PGLA-carbon fibres composite behavior under ‘in vivo’ condition [J]. Journal of Molecular Structure, 2008(875): 101−107.
[16] 曹伟伟, 朱 波, 井 敏, 等. PAN基碳纤维在表面处理中的拉曼光谱研究[J]. 光谱学与光谱分析, 2008, 28(12): 2885− 2889. CAO Wei-wei, ZHU Bo, JING Min, et al. Raman spectra of PAN-Based carbon fibers during surface treatment [J]. Spectroscopy and Spectral Analysis, 2008, 28(12): 2885−2889.
[17] SADEZKY A, MUCKENHUB H, GROTHE H, et al. Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information [J]. Carbon, 2005, 43(8): 1731−1742.
(编辑 高海燕)
Effect of HNO3oxidation treatment on surface components and structure of domestic PAN-based carbon fibers
LI Chan, WU Shuai, LIU Yun-qi, WU Huang, ZHAO Yun-hao, YI Mao-zhong, GE Yi-cheng
(State Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, China)
The domestic PAN-based carbon fiber was surface treated by nitric acid. The change of the surface active groups and microstructure were investigated by nitrogen -oxygen-hydrogen analyzer, X-ray photoelectron spectroscopy, Infrared spectrometry, Raman spectroscopy and Scanning Electron Microscope. The results indicate that the mass of carbon fiber decreases after the oxidation treatment at 55 ℃, but increases after treated at 80 ℃and 100 ℃. By carbon fibers surface oxidation treatment, the total and the external oxygen concentration increase, andthe overall content is significantly lower than the surface, while the growth is above the surface. The generation and transformation of —OH, C—O, C=O, COOR andpyridine moieties, quaternary nitrogen, oxidesed forms of nitrogen (—NO2) occured simultaneously, forming a dynamic process of groups transformation from low energy to high energy.With increasing oxidation temperature and time, the disorder degree of the carbon fibers surface decrease.
domestic PAN-based carbon fiber; surface oxidation; functional groups; microstructure
TB332
A
1673-0224(2015)2-250-08
国防项目
2014-03-21;
2014-05-04
易茂中,教授,博士。电话:0731-88877700;E-mail: yimaozhong@126.com
