谱系特有基因研究进展
2015-10-13张焕萍尹佟明
张焕萍,尹佟明
1.南京林业大学机电学院,南京 210037;
2.南京林业大学南方现代林业协同创新中心,南京 210037
谱系特有基因研究进展
张焕萍1,2,尹佟明2
1.南京林业大学机电学院,南京 210037;
2.南京林业大学南方现代林业协同创新中心,南京 210037
谱系特有基因(Lineage-specific genes,LSGs)是指在一个谱系中特有并与其他物种谱系所有基因没有明显序列相似性的基因,约为物种基因组全部基因数量的10%~20%,于1996年首次在完成全基因组测序的酵母基因组中大量发现。大规模测序技术的发展使谱系特有基因研究成为比较基因组学的研究热点,已在微生物、海洋低等生物、植物(如拟南芥、水稻、杨树)、昆虫及高等灵长类动物等多个物种或类群中展开,其生物功能对于阐明物种进化历程和生物适应性具有重要意义。文章介绍了谱系特有基因的研究背景和现状,从谱系特有基因获取、基因结构分析、进化起源、生物功能、表达特性分析等方面阐述谱系特有基因的研究进展,分析了存在的问题和后续研究方向,以期为相关研究提供参考。
谱系特有基因;孤儿基因;基因起源进化;基因生物功能
谱系特有基因(Lineage-specific genes,LSGs)又称为分类受限基因(Taxonomically-restricted genes, TRGs),是指在一个谱系中特有、与其他物种谱系所有基因没有明显序列相似性的基因[1],在相关文献中也称为非严格意义上的孤儿基因(Orphan genes)或ORFans(ORFans多用于微生物)[2]。1996年,最初在完成全基因组测序的酵母(Saccharomyces cerevisiae)基因组发现大量与数据库序列无相似性的孤儿基因,约为酵母基因组数量的26%[3]。随着测序技术的发展,越来越多的物种完成了全基因组测序,通过序列比对,研究人员在多个物种中发现了约为基因组数量10%~20%的谱系特有基因[4]。已完成测序的物种数量对谱系特有基因的比例和数量有很大影响(图1),用树状图表示测序物种数量对孤儿基因的影响,测序物种数量的增加使原来的孤儿基因变成谱系特有基因,在相关文献及本文中,对孤儿基因和谱系特有基因并不进行严格意义上的区分。
谱系特有基因与新基因这两个概念有一定的相关性,但并不等同。Long等[5]认为广义新基因(New gene)是指物种某一位点在进化过程中特定时间范畴内产生的此前不存在的基因。复制产生的新基因通常是多基因家族的成员,不能用序列相似性方法鉴定出来,因此多数新基因不是谱系特有基因,例如人类基因组在近期进化过程中产生了大量的新基因,只有一部分基因是人类特有的基因[6]。年轻基因(Young gene)由于产生时间短,保留了进化过程中大量有用的信息,这些信息对于新基因研究有重要作用,进化时间在几百万年的年轻新基因通常是谱系特有基因,目前大多数新基因研究都以年轻新基因为主[5]。Tautz等[7]研究结果表明,小鼠(Mus musculus)基因组中的孤儿基因主要是从头(de novo)起源新基因,即相当多数的谱系特有基因或孤儿基因可能都是新基因;谱系特有基因也可以是老的复制基因因为快速进化失去了与其同源基因的相似性而产生[8]。
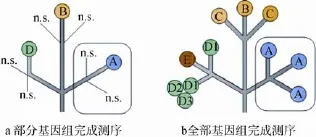
图1 测序物种数量对谱系特有基因鉴定的影响[4]a:9个物种分别代表3个属,3个属各有1个物种完成基因组测序,得到3个孤儿基因A、B、D,n.s.表示未测序的基因组;b:9个物种全部测序完成,基因B和基因E是严格意义上的孤儿基因,基因A是同属物种特有基因,基因D1、D2、D3旁系同源,基因C是同属物种特有基因。
谱系特有基因通常有相似的基因结构如肽链长度短、内含子数量少等,但起源机制、生物功能、表达水平及表达偏好性有各自的特性,在不同基因组中的比例也有差异,研究者通过对比多个物种,发现植物谱系特有基因平均比例高于动物中的平均比例[9]。谱系特有基因获取受到测序技术的影响,微生物中已有大量物种完成测序,因此谱系特有基因研究也相对深入。Yin等[10,11]对上千种微生物基因组进行比对,发现病毒基因组中约30%的开放阅读框 (Open reading frame,ORF)是ORFans,细菌基因组ORFans比例约为9.1%,表明ORFans在病毒中起源速率较高。高等生物由于完成测序的基因组数量少,谱系特有基因研究还不够深入,大多数谱系特有基因的生物功能是未知的,其起源进化也尚未完全研究清楚,但谱系特有基因的生物功能往往与物种特有的生物特征和环境适应性有关,这对于阐明物种进化历程有重要意义。目前谱系特有基因研究已成为比较基因组学的新兴研究领域,并在多个物种或类群中展开,如微生物[10~12]、高等灵长类动物[13~16]、昆虫[17~20]及拟南芥(Arabidopsis thaliana)[21,22]、水稻(Oryza sativa)[23]、杨树(Populus)[24]等多个模式植物[25],与自然界庞大的物种数量相比,谱系特有基因研究仍在起步阶段,还有大量工作要做。本文详细介绍了谱系特有基因的研究背景和现状,从谱系特有基因获取、基因结构分析、基因进化起源、生物功能预测、基因表达分析等方面阐述谱系特有基因的研究进展及取得的成果,分析了存在的问题和后续研究方向。
1 谱系特有基因获取方法
谱系特有基因的获取一般是通过序列比对,将所研究物种的测序序列(基因组序列、转录组序列等)与其他物种进行比对,比对物种序列主要从NCBI等多个数据库中获得,根据比对相似程度获得谱系特有基因,所得基因数量多少取决于谱系的定义范围,并受到比对序列和比对算法的影响。常用的比对软件以BLAST为主,包括 BLASTP、TBLASTN、PSIBLAST等,其他比对软件有liftOver、InterProScan[26]等。比对结果受数据库选取、比对算法、参数设置等因素的影响,不同研究小组获取的同一物种谱系特有基因数量也有差异,如灵长类动物和拟南芥。Tay等[13]用基于BLASTZ的软件liftOver将从cDNA和EST实验数据获得38037转录单元与9个非灵长类物种基因组进行比对,得到131个灵长类谱系特有转录单元,其结果与Toll-Riera等[14]对灵长类动物研究结果有差异。Lin等[21]比较了拟南芥与178种植物的基因序列,获得两组谱系特有基因,一组是914个仅与十字花科植物有序列相似的十字花科谱系特有基因,另一组是1324个与其他物种都没有序列相似的拟南芥谱系特有基因,这些基因大部分被转录,但没有基因功能注释;Donoghue等[22]用BLAST软件和InterProScan对数据库中的大量物种序列与拟南芥基因组序列进行比对筛选(包括线粒体基因和叶绿体基因),得到1789个(28个线粒体)十字花科谱系特有基因,其中958个(18个线粒体)拟南芥谱系特有基因,叶绿体中无十字花科谱系特有基因,与Lin等的研究结果有差异。这种差异表明用生物计算的方法获取谱系特有基因会受到计算参数的影响,具有较高的假阳性率。如何建立一种快速、有效的谱系特有基因鉴定方法,对后续研究有很大影响,同时各物种所获得的谱系特有基因也会随着基因组数据的增加、基因注释的改善而变化。目前谱系特有基因绝对数量在增长中,随着更多基因组测序的完成,会使原有的谱系特有基因在其他物种中找到同源基因,谱系特有基因数量会逐渐下降或趋于饱和。Wilson等[12]研究了最早发表的122种微生物中的孤儿基因,发现孤儿基因的比例随着测序基因组数量的增加而下降,但绝对数量还在小幅增长。
2 谱系特有基因结构特性分析
基因结构分析主要包括基因长度、内含子数量及长度、外显子数量及长度、GC含量、染色体分布偏好性等。多数物种的谱系特有基因具有相似的结构特征,如肽链长度短、外显子和内含子数量少、GC含量低等特性,灵长类动物、拟南芥、果蝇(Drosophila)、蜜蜂(Apis mellifera)、斑马鱼(Danio rerio)等谱系特有基因结构都有这种特性[14,17,22],如斑马鱼基因组中[9],约28%的孤儿基因只有一个外显子,而保守基因中只有一个外显子的基因比例是6%。玉米(Zea mays)基因组中约36.87%的基因是无内含子基因,其中约有1/6的无内含子基因与其他物种无同源性,这些基因对于研究玉米的进化起源和特有的生物功能有重要意义[27]。多数物种谱系特有基因GC含量低于保守基因,这一特性并不具有普遍性,例如斑马鱼中孤儿基因GC含量高于保守基因,这一特性与水稻谱系特有基因相似[9]。谱系特有基因在染色体上的分布特性也不相同,斑马鱼的谱系特有基因在染色体上分布不均匀,部分染色体上谱系特有基因比例较高,而有些染色体上没有谱系特有基因,这种分布特性可能与染色体长度有关[9];拟南芥谱
系特有基因在5条染色体的比例分别是5.84%、7.86%、6.87%、6.07%和6.36%,表明拟南芥谱系特有基因的分布没有染色体偏好性[22]。蚂蚁基因组中的孤儿基因没有集聚性,均匀分布在整个基因组非孤儿基因之间[28]。
Tautz等[7]认为谱系特有基因和孤儿基因的结构特征还与起源方式及进化时间有关,从头起源的年轻基因,由于进化时间不长,通常基因长度较短,而通过复制-分化产生的基因,基因长度与起源时间没有相关性,因为长基因与短基因的复制机率是相同的。从头起源的基因初始阶段没有结构域,与其他基因共享结构域,且外显子数量少,并在进化过程中获得结构域和新的外显子[29]。起源后的新基因如何获得调控转录因子,目前还知之甚少,可能是基因组中还有许多未发现的转录起始位点[30],也可能新基因会利用已有的调控因子[7]。目前谱系特有基因结构分析仅限于少数物种,还不能得出谱系特有基因结构特性的普遍规律,具有相似结构特性的基因可能会有相同的起源和进化模式,基因结构分析有助于阐明谱系特有基因的进化起源机制。
3 谱系特有基因起源进化
与具有上亿年的多物种分布的古老保守基因相比,通常在近缘物种分布的谱系特有基因进化时间还很短。谱系特有基因的起源进化机制已成为研究热点,多数研究结果表明,与古老保守基因或分布广泛的基因相比,谱系特有基因的进化速度较快,且这种进化模式存在于多个物种[31~33],Albà等[34]发现哺乳动物基因的年龄与进化速率成反比关系,这种快速进化模式产生的内在机制并不明确,已有多种学说来解释这种进化机制[35,36]。
Long等[37]系统介绍了新基因起源的分子机制,起源机制主要有如下几种:基因复制(G e n e du pl i c a t i o n,GD)、转位因子(Tr a n s p o s o n exaptation,TE)、横向基因转移(Horizontal gene transfer,HGT)、从头进化机制(De novo)等,祖先基因在不同物种中的选择性保留也是谱系特有基因起源的一个重要机制[38]。基因复制被认为是最主要的新基因产生机制,并根据重复区域大小,可将基因复制分为单个基因复制、部分基因组复制和整个基因组复制即多倍体化,基因复制观点认为谱系特有基因是由基因复制后序列变异形成的,由于进化速度加快,该基因失去了与其他物种基因的序列相似性,从而出现了谱系特有基因[39,40],即Tautz等[1]认为的复制-分化(Duplication-divergence)模型,拟南芥、水稻、果蝇及灵长类动物等物种谱系特有基因的产生多数是基于基因复制[14,22,41]。横向基因转移指不同于常规的由亲代到子代的垂直基因传递(Vertical gene transfer,VGT),而是在不同生物个体之间,或细胞内部细胞器之间所进行的遗传物质的交流。横向基因转移是原核生物基因组取得新基因的主要途径,受体生物通过获取外源基因迅速得到有利的表现型,例如细菌不仅能快速产生耐药性,而且能在菌落间相互传播耐药性[42];相对于原核生物,横向基因转移在真核生物尤其是多细胞真核生物中发生频率可能较低,但横向基因转移对于真核生物的早期进化、提高环境适应性、新基因的产生发挥了重要作用[43~47]。从头起源是新基因起源的重要机制之一,新基因可以起源于基因间的非编码区[48],也可以起源于内含子[49],已在多个物种中发现了从头起源的基因[1,7,50~52]。C a r v u n i s等[53]在酿酒酵母(Saccharomyces cerevisiae)非基因序列中发现数百个物种特异的短ORF翻译活动,从中确定了多个原基因(proto-genes),这些原基因进而可能成为新基因;从头起源对于果蝇新基因的出现发挥了重要作用[54],约有11.9%的果蝇新基因是从头起源;Toll-Riera等[14]发现62个灵长类动物谱系特有基因是基于从头起源;蚂蚁谱系特有基因产生的主要机制是从头起源[28]。基因重叠(Gene overprinting)是从头起源的特例,这种起源机制是在注释基因中产生新的重叠阅读框并转录、翻译,原基因生物功能同时保留[55],两个重叠基因通常其中一个是谱系特有基因,另一个分布广泛[56],基因重叠起源机制存在于多个物种并在微生物中大量出现[57~60]。转位因子也是谱系特有基因起源的重要机制,转位因子是基因组中具有转位特性的独立的DNA片段,它们可以直接从基因组内的一个位点移到另一个位点,拟南芥约有10%的基因起源于转位因子[61];Böhne等[62]研究了多种脊椎动物,发现转位因子是脊椎动物进化和保持生物多样性的重要机制。同一物种谱系特有基因有不同的起源机制,灵长类动物谱系特有基因主要有3种起源机制,分别是基因复制、转位因子机制、非编码区从头进化机制[14];约2/3的十字花科植物谱系特有基因已鉴定起源机制,主要有基因复制、转位因子机制和基因重叠等[22]。
新基因通常由多种起源机制共同作用产生,由基因复制产生的新基因可能经历固定阶段(Fixation phase)、命运决定阶段(Fate-determination phase)和保存阶段(Preservation phase)[63],并以很低的概率在群体中固定,多数新基因在进化过程中丢失,固定下来的基因可能成为假基因,也可能保留原功能或成为具有新功能的基因,并在不断进化中适应外界环境成为谱系特有的关键基因。已有多种基因进化的模型和假说,包括新功能化(Neofunctionalization)模型[64]、 复 制 -退 化 -互 补(Duplication-degeneration-complementation,DDC)模型[65]、自适应辐射(Adaptive radiation,AR)模型[66]、创 新 - 扩 增 - 分 化 (Innovationamplification-divergence,IAD)模型[67]、解除适应性冲突(Escape from adaptive conflict,EAC)模型[68]、统计检验方法等等[37,63,69],这些模型主要针对以基因复制起源的新基因。生物体由简单到复杂的进化过程正是伴随着新基因不断产生和老基因丢失的过程,目前人们对这个过程的研究仍在初始阶段,有许多问题还知之甚少,如新基因如何在群体中固定,基于多种起源机制的新基因如何进化,谱系特有新基因的进化模型和基因结构产生的分子机制,不同谱系的新基因起源进化模式是否相同等等,这些问题已成为基因起源进化的重要研究内容。
4 谱系特有基因生物功能研究
物种进化过程中出现的新基因曾被认为是不重要的,其功能的缺失并不会对生物体有致命的影响,只有保守基因对于生命的延续才有不可替代的重要作用[70,71]。新基因在快速进化过程中受到外界环境压力会获得新的生物功能,新功能使物种更能适应外界环境而保留下来,并使新基因在进化过程中成为重要基因,这种模型称为新基因进化的“等待模型”[72,73],等待过程可能无需太长时间,新基因可以很快获得重要功能。最近,芝加哥大学的研究结果表明新基因在物种生长发育过程中与古老保守基因同样重要,该课题组利用RNA干扰技术研究了黑腹果蝇195个起源于一百万到三百五十万年前的新基因,阻止每个目标基因转录为功能产物,发现59个基因可在果蝇中后期发育阶段导致死亡,而古老保守基因在发育早期具有重要作用[74];该研究还发现一些具有重要生物功能的新基因只存在于某些物种,并在特定组织中表达,例如新基因nsr(novel spermatogenesis regulator)和Zeus只存在于少数果蝇物种,已成为果蝇生殖系统发育的重要基因[75,76]。这些谱系特有的新基因如何在较短的进化时间内成为物种发育的重要基因以及这些基因的生物功能,还有待进一步研究[77]。
用生物实验方法对新基因生物功能进行分析之前,通常先预测新基因的生物功能,常用方法有ORF长度分析、基因转录分析、Ka/Ks比率[78]、多态性分析等[37]。由于与其他物种无同源性,谱系特有基因无法通过同源比对方法推测其生物功能,这也增加了谱系特有基因生物功能预测的难度,因此大多数谱系特有基因生物功能是未知的,例如灵长类动物谱系特有基因,除少数几个基因外,大多数基因的生物功能并不明确,但这些谱系特有基因的表达通常具有组织特异性[14]。研究者认为谱系特有基因的生物功能与物种独特的生物特征及环境适应性有关,许多细菌物种对人类肠道就有特殊的环境适应性,人类肠道中发现的微生物蛋白质家族在其他环境中并不存在[79]。水蚤(Daphnia pulex)基因组是首次获得的甲壳动物的完整基因组序列,这个看似简单的小生物包含了约31 000个基因,超过1/3的基因为水蚤谱系特有,且这些水蚤谱系特有基因(包括多个生物功能未注释基因)均对生态环境的改变非常敏感[80]。刺胞动物都具有用以捕食的刺细胞,但不同刺胞动物如水螅(Hydra)、珊瑚(Acropora)的刺细胞形状和大小也不相同,以适应不同的生存环境和食物,提高防御能力,谱系特有基因对于刺胞动物形态多样性的形成发挥了重要作用[81~83]。果蝇谱系特有的flightin基因能增强双翅的飞行力量,提高果蝇的生存适应性[84]。模式植物拟南芥谱系特有基因的生物功能也与环境适应能力有很大关系,在外界环境刺激(生物刺激或非生物刺激)下,这些基因往往有相似的表达特性[22,85]。
一些新基因起源后很快参与到生物网络和生物通路中并发挥重要作用,果蝇具有重要生物功能的新基因多数参与了蛋白质相互作用网络,一些新基因与原有基因有多个相互作用,并成为生物网络中的重要节点。Zeus基因在约4~6百万年前起源于DNA结合蛋白Caf40,保留了Caf40约30%的DNA结合位点,获得了193个新的结合位点,调控下游多个参与生殖系统的基因,表明新基因可以在很短的进化时间内改变原有的生物网络[76]。酵母新基因能以独特的方式获得生物功能并参与和改变原有的生物网络[86]。这些谱系特有的新基因如何能在很短的进化时间内参与到复杂的生物网络并发挥重要作用,以及新基因在生物网络中的具体功能,还有待深入研究。
5 谱系特有基因表达分析
谱系特有基因的表达特性在各物种并不相同,在生物体发育的不同阶段和不同组织中也有差异,相对于非谱系特有基因,谱系特有基因表达水平较低,在水稻、果蝇及灵长类动物中都具有这样的特性[14,41,87]。拟南芥谱系特有基因的表达与非谱系特有基因相比,具有表达水平低、组织特异性、易受外界压力影响等特点,在外界环境刺激下(如冷、热、干旱、盐碱、病菌处理等),谱系特有基因的表达水平有不同的变化[22]。表达特性分析有助于阐明谱系特有基因的生物功能,常用的基因表达分析方法有EST[88]、Microarray[89]、RNA-seq[90]等技术。
谱系特有基因在生物体发育的不同阶段具有不同的表达模式,种系特征性发育阶段(Phylotypic stage)是动物胚胎发育比较相似的阶段,分化通常发生在该阶段之前或之后[91,92]。Domazet-Lošo等[93,94]研究了斑马鱼和黑腹果蝇的基因在不同发育阶段的转录表达,发现这两种生物在种系特征性发育阶段有最保守的转录组,古老基因高表达和年轻基因低表达,而在之前的卵期和之后的成年期,则有较新的转录组,古老基因表达下降和年轻基因表达上升,表明斑马鱼和黑腹果蝇谱系特有的适应性特征在种系特征性发育阶段形成的可能性较小,这些适应性特征更可能是在之前的卵期和之后的成年期形成,这种发育模式称为“沙漏模式(Hourglass model)”,种系特征性发育阶段是发育调节和发育制约的关键点。多种动物胚胎发育过程中存在种系特征性发育阶段,大量保守基因在该阶段表达[95~97]。Quint等[98]研究发现,“沙漏模式”不仅存在于动物界,模式植物拟南芥中也存在相同的发育模式。
谱系特有基因的表达还具有组织差异性,多数谱系特有基因尤其是动物谱系特有基因通常在生殖系统和脑部这两类组织中特异表达。斑马鱼[9]、灵长类动物[13]、果蝇[87]的谱系特有基因在生殖系统都有较高表达,果蝇多个谱系特有新基因在生殖系统发育中发挥重要作用,如nsr和Zeus基因[75~76,99];黑腹果蝇(D.melanogaster)的谱系特有基因 Sdic1 (Sperm-specific dynein intermediate chain 1)在生殖器官中特异表达[100]。研究者应用染色体工程将果蝇(D.melanogaster)中Sdic基因家族所有拷贝去除,结果导致果蝇精子活力大大下降,表明Sdic基因家族与果蝇生殖能力相关[101,102];果蝇逆转座基因Drcd-1r控制生殖器官基因表达和雄性果蝇的繁殖[103]。由于动物生殖器官睾丸中有大量特异表达的新基因,Kaessmann等[72]提出了“out of testis”假说,该假说认为睾丸是促进动物基因组新基因起源和进化的重要器官。植物中新基因起源研究比较少,张亚平院士课题组利用基因芯片表达数据发现拟南芥中新基因在成熟花粉中显著偏好表达,提出了植物基因组新基因产生的“Out of pollen”假说[104]。多数物种谱系特有基因在脑部组织特异表达,果蝇多个谱系特有新基因如sphinx在脑部特异表达并控制果蝇行为[105,106];Fortna等[107]利用cDNA技术研究人类谱系特有基因,发现这些基因生物功能与脑部神经发育有关。Zhang等[108]研究发现,多个人类特有的年轻基因在脑部前额皮层高表达,其生物功能与认知活动相关,并在胎儿大脑中更为常见。人类特有的年轻基因SRGAP2在大脑神经发育中起关键作用,该基因缺失会造成神经连接发育出现问题,导致常见的神经发育疾病比如自闭症,癫痫和精神分裂症等[109,110]。Wu等[111]鉴定出60个从头起源的人类谱系特有新基因,RNA-seq数据显示这些基因在大脑皮层和睾丸组织中表达最高,表明这些基因的生物功能与人类认知能力有关。谱系特有基因在不同阶段和不同组织的表达特性,对于研究这些基因的生物功能和参与的生物过程具有重要意义。
6 结 语
自然界的物种在不断进化中以适应变化的生存环境,谱系特有基因的产生为物种的生物适应性和遗传多样性提供了物质基础。虽然谱系特有基因研究取得了一些成果,但研究范围还局限于少数物种,整体研究仍在起步阶段,对于谱系特有基因的进化起源、生物功能等方面还知之甚少,仍有许多问题没有解决,例如:(1)利用基因组比对的方法鉴别谱系特有基因受到计算方法的很大影响,建立一套高效的谱系特有基因鉴定方法是待解决的问题之一;(2)谱系特有基因的基因结构与起源机制、起源动力的关系,以及基因结构对生物功能和表型的影响;(3)谱系特有基因如何参与到生物体复杂的生物网络并行使生物功能,如何参与生物体重要组织的生命活动;(4)不同物种谱系特有基因的横向对比研究,总结谱系特有基因起源进化规律和生物功能。大规模测序技术的发展使许多物种完成了基因组测序,不断有新的基因组序列被公布,越来越多的近缘物种测序产生了大量数据,从中将能找到关于谱系特有基因起源与进化的有用信息,为谱系特有基因的全面深入研究提供了有利条件,这对于阐明物种进化历程和生物适应性具有重要意义。
[1]Tautz D,Domazet-Lošo T.The evolutionary origin of orphan genes.Nat Rev Genet,2011,12(10):692–702.
[2]Fischer D.Eisenberg D.Finding families for genomic ORFans.Bioinformatics,1999,15(9):759–762.
[3]Dujon B.The yeast genome project:what did we learn? Trends Genet,1996,12(7):263–270.
[4]Khalturin K,Hemmrich G,Fraune S,Augustin R,Bosch TCG.More than just orphans:are taxonomically-restricted genes important in evolution?Trends Genet,2009,25(9): 404–413.
[5]Chen SD,Krinsky BH,Long MY.New genes as drivers of phenotypic evolution.Nat Rev Genet,2013,14(9):645–660.
[6]Zhang YE,Landback P,Vibranovski MD,Long MY. Accelerated recruitment of new brain development genes into the human genome.PLoS Biol,2011,9(10): e1001179.
[7]Neme R,Tautz D.Phylogenetic patterns of emergence of new genes support a model of frequent de novo evolution. BMC Genomics,2013,14:117.
[8]Magadum S,BanerjeeU,MuruganP,GangapurD, Ravikesavan R.Gene duplication as a major force in evolution.J Genet,2013,92(1):155–161.
[9]YangLD,ZouM,FuBD,HeSP.Genome-wide identification,characterization,and expression analysis of lineage-specific genes within zebrafish.BMC Genomics, 2013,14:65.
[10]Yin YB,Fischer D.Identification and investigation of ORFans in the viral world.BMC Genomics,2008,9:24.
[11]Yin YB,Fischer D.On the origin of microbial ORFans: Quantifying the strength of the evidence for viral lateral transfer.BMC Evol Biol,2006,6:63.
[12]Wilson GA,Bertrand N,Patel Y,Hughes JB,Feil EJ,Field D.Orphans as taxonomically restricted and ecologically important genes.Microbiology,2005,151(8):2499–2501.
[13]Tay SK,Blythe J,Lipovich L.Global discovery of primate-specific genes in the human genome.Proc Natl Acad Sci USA,2009,106(29):12019–12024.
[14]Toll-Riera M,Bosch N,Bellora N,Castelo R,Armengol L, Estivill X,Albà MM.Origin of primate orphan genes: a comparative genomics approach.Mol Biol Evol,2009, 26(3):603–612.
[15]Lindskog C,Kuhlwilm M,Davierwala A,Fu N,Hegde G, UhlénM,NavaniS,Pääbo S,Pontén F.Analysis of candidate genes for lineage-specific expression changes in humansand primates.J Proteome Res,2014,13(8): 3596–3606.
[16]Zhang Q. Using pseudogene database to identify lineage-specific genes and pseudogenes in humans and chimpanzees.J Hered,2014,105(3):436–443.
[17]Johnson BR,Tsutsui ND.Taxonomically restricted genes are associated with the evolution of sociality in the honey bee.BMC Genomics,2011,12:164.
[18]Zhang GJ,Wang HS,Shi JJ,Wang XL,Zheng HK,Wong GKS,Clark T,Wang W,Wang J,Kang L.Identification and characterization of insect-specific proteins by genome data analysis.BMC Genomics,2007,8:93.
[19]Palmieri N,Kosiol C,Schlötterer C.The life cycle of Drosophila orphan genes.Elife,2014,3:e01311.
[20]Rogers RL,Shao L,Sanjak JS,Andolfatto P,Thornton KR. Revised annotations, sex-biased expression, and lineage-specific genes in the Drosophila melanogaster group.G3(Bethesda),2014,4(12):2345-2351.
[21]Lin HN,Moghe G,Ouyang S,Iezzoni A,Shiu SH,Gu X, Buell CR.Comparative analyses reveal distinct sets of lineage-specific genes within Arabidopsis thaliana.BMC Evol Biol,2010,10:41.
[22]Donoghue MT,Keshavaiah C,Swamidatta SH,Spillane C. Evolutionary origins of Brassicaceae specific genes in Arabidopsis thaliana.BMC Evol Biol,2011,11:47.
[23]Campbell MA,Zhu W,Jiang N,Lin HN,Ouyang S,Childs KL,Haas BJ,Hamilton JP,Buell CR.Identification and characterization of lineage-specific genes within the poaceae.Plant Physiol,2007,145(4):1311–1322.
[24]Lin WL, Cai B, Cheng ZM. Identification and characterization of lineage-specific genes in Populus trichocarpa.Plant Cell Tiss Org,2014,116(2):217–225.
[25]Yang XH,Jawdy S,TschaplinskiTJ,Tuskan GA. Genome-wide identification of lineage-specific genes in Arabidopsis,Oryza and Populus.Genomics,2009,93(5): 473–480.
[26]Hunter S,Apweiler R,Attwood TK,Bairoch A,Bateman A,Binns D,Bork P,Das U,Daugherty L,Duquenne L, Finn RD,Gough J,Haft D,Hulo N,Kahn D,Kelly E, Laugraud A,Letunic I,Lonsdale D,Lopez R,Madera M, Maslen J,McAnulla C,McDowall J,Mistry J,Mitchell A, Mulder N,Natale D,Orengo C,Quinn AF,Selengut JD, Sigrist CJA,Thimma M,Thomas PD,Valentin F,Wilson D,Wu CH,Yeats C.InterPro:the integrative protein signature database.Nucleic Acids Res,2009,37(suppl.1): D211–D215.
[27]Yan HW,Zhang W,Lin YX,Dong Q,Peng XJ,Jiang HY, Zhu SW,Cheng BJ.Different evolutionary patterns among intronless genes in maize genome.Biochem Biophys Res Commun,2014,449(1):146–150.
[28]WisslerL,Gadau J,Simola DF,HelmkampfM, Bornberg-Bauer E.Mechanisms and dynamics of orphan gene emergence in insect genomes.Genome Biol Evol, 2013,5(2):439–455.
[29]Chothia C,Gough J.Genomic and structural aspects of protein evolution.Biochem J,2009,419(1):15–28.
[30]Carninci P.RNA dust:where are the genes?DNA Res, 2010,17(2):51–59.
[31]Cai JJ,Woo PCY,Lau SKP,Smith DK,Yuen KY. Accelerated evolutionary rate may be responsible for the emergence of lineage-specific genes in ascomycota.J Mol Evol,2006,63(1):1–11.
[32]Wolf YI,Novichkov PS,Karev GP,Koonin EV,Lipman DJ.The universal distribution of evolutionary rates of genes and distinct characteristics of eukaryotic genes of different apparent ages.Proc Natl Acad Sci USA,2009, 106(18):7273–7280.
[33]Kasuga T,Mannhaupt G,Glass NL.Relationship between phylogenetic distribution and genomic features in Neurospora crassa.PLoS One,2009,4(4):e5286.
[34]Albà MM,Castresana J.Inverse relationship between evolutionary rate and age of mammalian genes.Mol Biol Evol,2005,22(3):598–606.
[35]Cai JJ,Petrov DA.Relaxed purifying selection and possibly high rate of adaptation in primate lineage-specific genes.Genome Biol Evol,2010,2:393–409.
[36]Gayà-Vidal M,Albà MM.Uncovering adaptive evolution in the human lineage.BMC Genomics,2014,15:599.
[37]Long MY,Van Kuren NW,Chen SD,Vibranovski MD. New gene evolution:little did we know.Annu Rev Genet, 2013,47:307–333.
[38]Guo LH,Chen YN,Ye N,Dai XG,Yang WX,Yin TM. Differential retention and expansion of the ancestral genes associated with the paleopolyploidies in modern rosid plants,as revealed by analysis of the extensins super-gene family.BMC Genomics,2014,15:612.
[39]Hoffmann FG,OpazoJC,StorzJF.Rapid ratesof lineage-specific gene duplication and deletion in the alpha-globin gene family. Mol Biol Evol, 2008, 25(3):591-602.
[40]Kondrashov FA.Gene duplication as a mechanism of genomic adaptation to a changing environment.Proc Biol Sci,2012,279(1749):5048–5057.
[41]Guo WJ, Li P, Ling J, Ye SP. Significant comparative characteristics between orphan and nonorphan genes in the rice(Oryza sativa L.)genome. Comp Funct Genomics,2007:21676.
[42]Davies J,Davies D.Origins and evolution of antibiotic resistance.Microbiol Mol Biol Rev,2010,74(3):417–433.
[43]Dunning Hotopp JC. Horizontal gene transfer between bacteria and animals.Trends Genet,2011, 27(4):157–163.
[44]Gao CH,Ren XD,Mason AS,Liu HL,Xiao ML,Li JN,Fu DH.Horizontal gene transfer in plants.Funct Integr Genomics,2014,14(1):23–29.
[45]Huang JL.Horizontal gene transfer in eukaryotes:The weak-link model.BioEssays,2013,35(10):868–875.
[46]王洽,乐霁培,张体操,黄锦岭,孙航.水平基因转移在生物进化中的作用.科学通报,2014,59(21):2055–2064.
[47]Cooper ED. Horizontal gene transfer: accidental inheritance drives adaptation.Curr Biol,2014,24(12): R562–R564.
[48]Cai J,Zhao RP,Jiang HF,Wang W.De novo origination of a new protein-coding gene in Saccharomyces cerevisiae. Genetics,2008,179(1):487–496.
[49]Sorek R.The birth of new exons:mechanisms and evolutionary consequences. RNA, 2007, 13(10): 1603–1608.
[50]Light S,Basile W,Elofsson A.Orphans and new gene origination,a structural and evolutionary perspective. Curr Opin Struct Biol,2014,26:73–83.
[51]Wu DD,Zhang YP.Evolution and function of de novo originated genes. Mol Phylogenet Evol, 2013, 67(2):541–545.
[52]Li CY,Zhang Y,Wang ZB,Zhang Y,Cao CM,Zhang PW, Lu SJ,Li XM,Yu Q,Zheng XF,Du Q,Uhl GR,Liu QR, Wei LP.A human-specific De novo protein-coding gene associated with human brain functions.PLoS Comput Biol, 2010,6(3):e1000734.
[53]Carvunis AR,Rolland T,Wapinski I,Calderwood MA, Yildirim MA,Simonis N,Charloteaux B,Hidalgo CA, Barbette J,Santhanam B,Brar GA,Weissman JS,Regev A, Thierry-Mieg N,Cusick ME,Vidal M.Proto-genes and de novo gene birth.Nature,2012,487(7407):370–374.
[54]Zhou Q,Zhang GJ,Zhang Y,Xu SY,Zhao RP,Zhan ZB, Li X,Ding Y,Yang S,Wang W.On the origin of new genes in Drosophila.Genome Res,2008,18(9):1446–1455.
[55]Ohno S.Birth of a unique enzyme from an alternative reading frame of the preexisted,internally repetitious coding sequence.Proc Natl Acad Sci USA.1984,81(8):2421–2425.
[56]KeesePK,Gibbs A.Originsofgenes:"big bang" or continuous creation?Proc Natl Acad Sci USA,1992, 89(20):9489–9493.
[57]Ekman D,Elofsson A.Identifying and quantifying orphan protein sequences in fungi.J Mol Biol,2010,396(2): 396–405.
[58]ChungWY,WadhawanS,SzklarczykR,PondSK, Nekrutenko A.A first look at ARFome:Dual-coding genes in mammalian genomes.PLoS Comput Biol,2007,3(5): e91.
[59]Knowles DG,McLysaght A.Recent de novo origin of human protein-coding genes.Genome Res,2009,19(10): 1752–1759.
[60]Sabath N,Wagner A,Karlin D.Evolution of viral proteins originated de novo by overprinting.Mol Biol Evol,2012, 29(12):3767–3780.
[61]Lockton S,Gaut BS.The contribution of transposable elements to expressed coding sequence in Arabidopsis thaliana.J Mol Evol,2009,68(1):80–89.
[62]Böhne A,Brunet F,Galiana-Arnoux D,Schultheis C,Volff JN. Transposable elements as drivers of genomic and biological diversity in vertebrates.Chromosome Res, 2008,16(1):203–215.
[63]Innan H,Kondrashov F.The evolution of gene duplications: classifying and distinguishing between models.Nat Rev Genet,2010,11(2):97–108.
[64]Gibson TA,Goldberg DS.Questioning the ubiquity of neofunctionalization.PLoS ComputBiol,2009,5(1): e1000252.
[65]Van Hoof A.Conserved functions of yeast genes support the duplication,degeneration and complementation model for gene duplication.Genetics,2005,171(4):1455–1461.
[66]Gillespie RG.Adaptive radiation: convergence and non-equilibrium.Curr Biol,2013,23(2):R71–R74.
[67]Näsvall J,Sun L,Roth JR,Andersson DI.Real-time evolution of new genes by innovation,amplification,and divergence.Science,2012,338(6105):384–387.
[68]Deng C,Cheng CHC,Ye H,He XM,Chen LB.Evolution of an antifreeze protein by neofunctionalization under escape from adaptive conflict.Proc Natl Acad Sci USA, 2010,107(50):21593–21598.
[69]孙红正,葛颂.重复基因的进化——回顾与进展.植物学报,2010,45(1):13–22.
[70]Krylov DM,Wolf YI,Rogozin IB,Koonin EV.Gene loss, protein sequence divergence, gene dispensability, expression level,and interactivity are correlated in eukaryotic evolution. Genome Res, 2003, 13(10): 2229–2235.
[71]Gabor Miklos GL,Rubin GM.The role of the genome project in determining gene function:insights from model organisms.Cell,1996,86(4):521–529.
[72]Kaessmann H.Origins,evolution,and phenotypic impact of new genes.Genome Res,2010,20(10):1313–1326.
[73]Long MY,Betrán E,Thornton K,Wang W.The origin of new genes:glimpses from the young and old.Nat Rev Genet,2003,4(11):865–875.
[74]Chen SD,Zhang YE,Long MY.New genes in Drosophila quickly become essential.Science,2010,330(6011): 1682–1685.
[75]Ding Y,Zhao L,Yang S,Jiang Y,Chen Y,Zhao RP,Zhang Y,Zhang GJ,Dong Y,Yu HJ,Zhou Q,Wang W.A young Drosophila duplicate gene plays essential roles in spermatogenesisby regulating severaly-linked male fertility genes.PLoS Genet,2010,6(12):e1001255.
[76]Chen SD,Ni XC,Krinsky BH,Zhang YE,Vibranovski MD,White KP,Long MY.Reshaping of global gene expression networks and sex-biased gene expression by integration of a young gene.EMBO J,2012,31(12): 2798–809.
[77]Cardoso-Moreira M,Long MY.The origin and evolution of new genes.Methods Mol Biol,2012,856:161–186.
[78]Nei M,Gojobori T.Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions.Mol Biol Evol,1986,3(5):418–426.
[79]Ellrott K,Jaroszewski L,Li WZ,Wooley JC,Godzik A. Expansion of the protein repertoire in newly explored environments:human gut microbiome specific protein families.PLoS Comput Biol,2010,6(6):e1000798.
[80]Colbourne JK,Pfrender ME,Gilbert D,Thomas WK, Tucker A,Oakley TH,Tokishita S,Aerts A,Arnold GJ, Basu MK,Bauer DJ,Cáceres CE,Carmel L,Casola C, Choi JH,Detter JC,Dong QF,Dusheyko S,Eads BD, Fröhlich T,Geiler-Samerotte KA,Gerlach D,Hatcher P, Jogdeo S,Krijgsveld J,Kriventseva EV,Kültz D,Laforsch C,Lindquist E,Lopez J,Manak JR,Muller J,Pangilinan J, Patwardhan RP,Pitluck S,Pritham EJ,Rechtsteiner A, Rho M,Rogozin IB,Sakarya O,Salamov A,Schaack S, Shapiro H,Shiga Y,Skalitzky C,Smith Z,Souvorov A, Sung W,Tang ZJ,Tsuchiya D,Tu H,Vos H,Wang M, Wolf YI,Yamagata H,Yamada T,Ye YZ,Shaw JR, Andrews J,Crease TJ,Tang HX,Lucas SM,Robertson HM,Bork P,Koonin EV,Zdobnov EM,Grigoriev IV, Lynch M,BooreJL.Theecoresponsivegenomeof Daphnia pulex.Science,2011,331(6017):555–561.
[81]Milde S.Hemmrich G,Anton-Erxleben F,Khalturin K, Wittlieb J,Bosch TCG.Characterization of taxonomically restricted genes in a phylum-restricted cell type.Genome Biol,2009,10(1):R8.
[82]Steele RE,David CN,Technau U.A genomic view of 500 million years of cnidarian evolution.Trends Genet,2011, 27(1):7–13.
[83]Khalturin K,Anton-Erxleben F,Sassmann S,Wittlieb J, Hemmrich G,Bosch TCG.A novel gene family controls species-specific morphological traits in Hydra.PLoS Biol, 2008,6(11):e278.
[84]Domazet-Loso T,Tautz D.An evolutionary analysis of orphan genes in Drosophila.Genome Res,2003,13(10): 2213–2219.
[85]Silverstein KAT,Moskal WA Jr,Wu HC,Underwood BA, Graham MA, Town CD, VandenBosch KA. Smallcysteine-rich peptidesresembling antimicrobial peptides have been under-predicted in plants.Plant J, 2007,51(2):262–280.
[86]Capra JA,Pollard KS,Singh M.Novel genes exhibit distinct patterns of function acquisition and network integration.Genome Biol,2010,11(12):R127.
[87]Begun DJ,Lindfors HA,Kern AD,Jones CD.Evidence for de novo evolution of testis-expressed genes in the Drosophila yakuba/Drosophila erecta clade.Genetics, 2007,176(2):1131–1137.
[88]Parkinson J,Blaxter M.Expressed sequence tags:an overview.Methods Mol Biol,2009,533:1–12.
[89]Kogenaru S,Qing Y,Guo YP,Wang NA.RNA-seq and microarray complementeach otherin transcriptome profiling.BMC Genomics,2012,13:629.
[90]祁云霞,刘永斌,荣威恒.转录组研究新技术: RNA-Seq及其应用.遗传,2011,33(11):1191–1202.
[91]Richardson MK.A phylotypic stage for all animals?Dev Cell,2012,22(5):903–904.
[92] Švorcová J.The phylotypic stage as a boundary of modular memory:non mechanistic perspective.Theory Biosci,2012,131(1):31–42.
[93]Domazet-Lošo T,Tautz D.A phylogenetically based transcriptome age index mirrors ontogenetic divergence patterns.Nature,2010,468(7325):815–818.
[94]Kalinka AT,Varga KM,Gerrard DT,Preibisch S,Corcoran DL,Jarrells J,Ohler U,Bergman CM,Tomancak P.Gene expression divergence recapitulates the developmental hourglass model.Nature,2010,468(7325):811–814.
[95]Hazkani-Covo E,Wool D,Graur D.In search of the vertebrate phylotypic stage:a molecular examination of the developmental hourglass model and von Baer's third law.J Exp Zool B Mol Dev Evol,2005,304(2):150–158.
[96]Irie N,Kuratani S.Comparative transcriptome analysis reveals vertebrate phylotypic period during organogenesis. Nat Commun,2011,2:248.
[97]Comte A,RouxJ,Robinson-RechaviM.Molecular signaling in zebrafish development and the vertebrate phylotypic period.Evol Dev,2010,12(2):144–156.
[98]Quint M,Drost HG,Gabel A,Ullrich KK,Bönn M, Grosse I. A transcriptomic hourglass in plant embryogenesis.Nature,2012,490(7418):98–101.
[99]Levine MT,Jones CD,Kern AD,Lindfors HA,Begun DJ. Novel genes derived from noncoding DNA in Drosophila melanogaster are frequently X-linked and exhibit testis-biased expression.Proc Natl Acad Sci USA,2006, 103(26):9935–9939.
[100]Nurminsky DI,Nurminskaya MV,De Aguiar D,Hartl DL. Selective sweep of a newly evolved sperm-specific gene in Drosophila.Nature,1998,396(6711):572–575.
[101]Yeh SD,Do T,Abbassi M,Ranz JM.Functional relevance of the newly evolved sperm dynein intermediate chain multigenefamily in Drosophila melanogastermales. Commun Integr Biol,2012,5(5):462–465.
[102]Yeh SD,Do T,Chan C,Cordova A,Carranza F,Yamamoto EA,Abbassi M,Gandasetiawan KA,Librado P,Damia E, DimitriP,RozasJ,HartlDL,RooteJ,RanzJM. Functional evidence that a recently evolved Drosophila sperm-specific gene boosts sperm competition.Proc Natl Acad Sci USA,2012,109(6):2043–2048.
[103]Quezada-Díaz JE,Muliyil T,Río J,Betrán E.Drcd-1 related:a positively selected spermatogenesis retrogene in Drosophila.Genetica,2010,138(9–10):925–937.
[104]Wu DD,Wang X,Li Y,Zeng L,Irwin DM,Zhang YP. "Out of pollen"hypothesis for origin of new genes in flowering plants:study from Arabidopsis thaliana. Genome Biol Evol,2014,6(10):2822–2829.
[105]Chen SD,Spletter M,Ni XC,White KP,Luo LQ,Long MY.Frequent recent origination of brain genes shaped the evolution of foraging behavior in Drosophila.Cell Rep, 2012,1(2):118–132.
[106]Chen Y,Dai HZ,Chen SD,Zhang LY,Long MY.Highly tissue specific expression of Sphinx supports its male courtship related role in Drosophila melanogaster. PLoS One,2011,6(4):e18853.
[107]Fortna A,Kim Y,MacLaren E,Marshall K,Hahn G, Meltesen L, Brenton M, Hink R, Burgers S, Hernandez-Boussard T,Karimpour-Fard A,Glueck D, McGavran L, Berry R, Pollack J, Sikela JM. Lineage-specific gene duplication and loss in human and great ape evolution.PLoS Biol,2004,2(7):E207.
[108]Zhang YE,Landback P,Vibranovski M,Long MY.New genes expressed in human brains:implications for annotating evolving genomes.Bioessays,2012,34(11): 982–991.
[109]Charrier C,Joshi K,Coutinho-Budd J,Kim JE,Lambert N, de Marchena J,Jin WL,Vanderhaeghen P,Ghosh A,Sassa T,Polleux F.Inhibition of SRGAP2 function by its human-specific paralogs induces neoteny during spine maturation.Cell,2012,149(4):923–935.
[110]Dennis MY,Nuttle X,Sudmant PH,Antonacci F,Graves TA,Nefedov M,Rosenfeld JA,Sajjadian S,Malig M, Kotkiewicz H,Curry CJ,Shafer S,Shaffer LG,de Jong PJ, Wilson RK,Eichler EE.Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication.Cell,2012,149(4):912–922.
[111]Wu DD,Irwin DM,Zhang YP.De novo origin of human protein-coding genes.PLoS Genet,2011,7(11):e1002379.
(责任编委:吴为人)
Advances in lineage-specific genes
Huanping Zhang1,2,Tongming Yin2
1.College of Mechanical and Electrical Engineering,Nanjing Forestry University,Nanjing 210037,China;
2.Co-Innovation Center for Sustainable Forestry in Southern China,Nanjing Forestry University,Nanjing 210037,China
Lineage-specific genes(LSGs)are defined as genes found in one particular taxonomic group but have no significant sequence similarity with genes from other lineages,which compose about 10%?20%of the total genes in the genome of a focal organism.LSGs were first uncovered in the yeast genome in 1996.The development of the whole genome sequencing leads to the emergence of studies on LSGs as a hot topic in comparative genomics.LSGs have been extensively studied on microbial species,lower marine organisms,plant(such as Arabidopsis thaliana, Oryza sativa,Populus),insects,primate,etc;the biological functions of LSGs are important to clarify the evolution and adaptability of a species.In this review,we summarize the progress of LSGs studies,including LSGs identification,gene characterization,origin and evolution of LSGs,biological function,and expression analysis of LSGs.In addition,we discuss the existing problems and future directions for studies in this area.Our purpose is to provide some unique insights into the researches of LSGs.
lineage-specific genes;orphan genes;origin and evolution of LSGs;biological function of LSGs
URL:http://www.cnki.net/kcms/detail/11.1913.R.20150316.1049.002.html
2014-11-13;
2015-02-20
行业公益重大项目(编号:201304102),国家自然科学基金项目(编号:31125008)和中国博士后基金项目(编号:2014M551604)资助
张焕萍,博士,讲师,研究方向:基因组学。E-mail:nuaazhp@njfu.edu.cn
尹佟明,博士,教授,博士生导师,研究方向:基因组学。E-mail:tmyin@njfu.edu.cn
10.16288/j.yczz.14-392
时间:2015-3-16 10:49:32
