1-(6-溴-3-吡啶基)-1-乙酮的合成研究
2015-09-07喻雅婷郭秀秀申凰林
曾 竟, 喻雅婷, 郭秀秀, 申凰林
(新疆师范大学化学化工学院,新疆乌鲁木齐830054)
1-(6-溴-3-吡啶基)-1-乙酮的合成研究
曾 竟, 喻雅婷, 郭秀秀, 申凰林
(新疆师范大学化学化工学院,新疆乌鲁木齐830054)
以2,5-二溴吡啶为原料,经过镁卤交换和亲核取代两步反应制得1-(6-溴-3-吡啶基)-1-乙酮化合物;反应主要以2 mo1%CuC1、2mo1%LiC1和2 mo1%A1C13为路易酸催化剂,在零摄氏度与室温之间,2-溴-5-吡啶基氯化镁与乙酰氯发生取代反应合成得到1-(6-溴-3-吡啶基)-1-乙酮,总收率高达81%。此合成方法具有原料来源方便、条件温和、收率高等优点。
2,5-二溴吡;镁卤交换;亲核取代反应;1-(6-溴-3-吡啶基)-1-乙酮
吡啶及其衍生物统称为吡啶碱,是医药、农药、合成材料、香料、染料等精细化学品的重要基础原料或中间体。目前应用范围很广,特别涉及医药[1,2]、农药[3]和饲料原料、新型高分子材料等许多重要领域。有文献报道的Mark.Cushman等人合成的取代吡啶基噻唑化合物能够显著抑制癌细胞的增殖[4]。而1-(6-溴-3 -吡啶基)-1-乙酮是合成此类化合物的关键中间体。此吡啶衍生物不仅酮羰基能发生亲核、氧化等有机化学反应,吡啶环上的卤原子也可被其它的原子或基团所取代衍生出更多的吡啶化合物,因此此类化合物的合成也引起人们关注,到目前为止,合成此类化合物主要包括两种方法:一是以6-溴烟酸为原料,通过与甲基镁试剂发生亲核取代反应而得到[4],但该路线中,使用原料价格昂贵,反应成本较高。除此之外,也有文献报道从2,5-二溴吡啶出发,在-78oC下与丁基锂发生锂卤交换后衍生而得到目标化合物[5],该路线原料便宜易得,但反应条件苛刻,收率中等,仅只有50%左右。
文章采用价格低廉的2,5-二溴吡啶为原料,在零摄氏度到室温之间,经过镁卤交换、亲核取代两步反应合成得到1-(6-溴-3-吡啶基)-1-乙酮,反应收率高达81%,与之前报道的合成路线相比具有原料来源方便、反应条件更加温和,产率较高等优点。合成路线具体如下:

图1 1-(6-溴-3-吡啶基)-1-乙酮的合成路线与结构
1 实验部分
1.1 试剂与仪器
镁屑、异丙基氯、2,5-二溴吡啶为工业级,1,2-二氯乙烷、乙酰氯、氯化亚铜、无水氯化锂、无水三氯化铝、四氢呋喃为分析纯。
X-4显微熔点仪(上海精密科学仪器有限公司);Nico1et-460型红外吸收光谱仪(美国Nico1et公司);AV500核磁共振波谱仪(瑞士Bruker公司)。
1.2 合成方法与表征
1.2.1 2-溴-5-吡啶基氯化镁(2)的制备
氮气保护下,将35.5 g(0.15mo1)的2,5-二溴吡啶(1)置于250mL的干燥四口瓶中,同时加入60mL无水四氢呋喃,启动搅拌,冰盐浴将反应体系降温至0oC左右,缓慢滴入65mL 2.5 M的异丙基氯化镁的四氢呋喃溶液,滴加过程中控制体系温度不超过10oC,异丙基氯化镁滴加完毕后缓慢回至室温,TLC跟踪反应直至镁卤交换完全得到2-溴-5-吡啶基氯化镁,直接留作下一步待用。
1.2.3 1-(6-溴-3-吡啶基)-1-乙酮(3)的合成与表征
氮气保护下,将0.28 g CuC1(2 mo1%),0.13 g LiC1(2 mo1%),0.41 g A1C13(2 mo1%)以及11.78 g(0.15 mo1)乙酰氯置于250mL的干燥四口瓶中,并用20mL无水四氢呋喃溶解,启动搅拌,反应体系用冰盐浴降温至0oC左右,从滴液漏斗中缓慢滴入上述镁卤交换得到的2的四氢呋喃溶液,滴加过程中控制体系温度不超过10oC,滴完后缓慢回至室温,TLC跟踪反应至结束,待反应完成后将反应体系倒入1M的NH4C1溶液中,并用二氯甲烷多次萃取有机相,合并有机相,有机相分别用碳酸氢钠溶液,饱和氯化钠溶液,蒸馏水洗涤,干燥后过滤并浓缩得粗品27.6 g,石油醚:乙酸乙酯 =5:1(V/V)重结晶得白色固体纯品24.3 g,产率81.0%。M.p.128-129oC(1it.,[6]127-129oC)。IR(CM-1,KBr):3093,1680,1578,1553,1104,839;1HNMR(400 MHz,CDC13)δ(ppm)2.63(s,3H),7.61(d,J=8.28Hz,1H),8.07-8.10(m,1H),8.90(d,J= 2.12Hz,1H)。
2 结果与讨论
2.1 6-溴-3-吡啶基氯化镁(2)的合成
本实验设计中第一步反应是镁卤交换,为放热反应。由于2,5-二溴吡啶中吡啶环上有两个不同位置的溴原子,为保证异丙基氯化镁能选择性地与5-位吡啶溴原子进行镁卤交换,最关键的是在无水条件下控制反应温度。在本设计中一方面采取在滴入异丙基氯化镁之前将整个反应体系降温至零摄氏度左右,保证开始滴入异丙基氯化镁时升温起点较低,另一方面则是控制异丙基氯化镁滴入速度为每秒1滴的速度,以此保证在滴加过程中体系温度不超过10oC。
2.2 1-(6-溴-3-吡啶基)-1-乙酮(3)的合成
2.2.1 催化剂用量对3收率的影响
6-溴-3-吡啶基氯化镁(2)与乙酰氯的反应为亲核取代反应,路易斯酸能促进此类反应。文章选用无水氯化亚铜、无水氯化锂和无水三氯化铝(摩尔比 =1:1:1)为路易斯酸催化剂并考察它们的用量对3收率的影响,结果如表1所示:
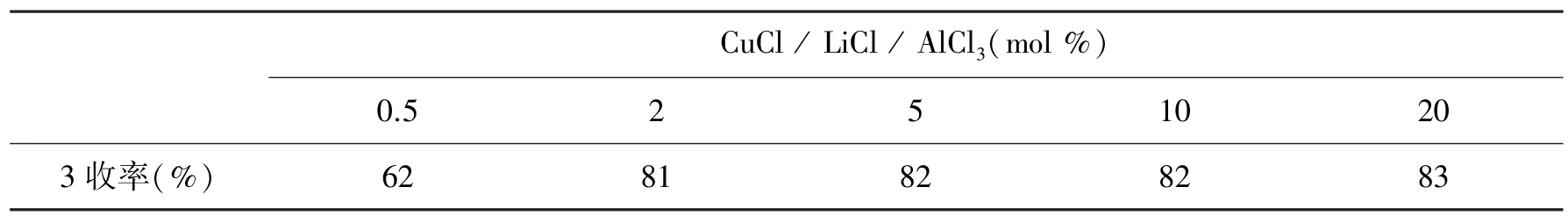
表1 无水氯化亚铜、无水氯化锂和无水三氯化铝(摩尔比 =1:1:1)为路易斯酸催化剂并考察它们的用量对3收率的影响
由表1所知,当催化剂用量为2 mo1%时,反应收率高达81%,而催化剂用量继续增加并没有明显提高3的产率,但当催化剂用量降至0.5 mo1%时,反应收率显著下降,仅只有62%。故确定选用2 mo1%CuC1、2 mo1%LiC1、2 mo1%A1C13为路易酸催化剂完成此亲核取代反应。
2.3 亲核取代反应反应温度对3收率的影响。
由于6-溴-3-吡啶基氯化镁(2)与乙酰氯的反应同样也是放热反应,并且生成产物3为活泼酰基化合物,对6-溴-3-吡啶基氯化镁较为敏感,因此也考察了反应温度对3收率的影响,实验结果表2所示:

表2 6-溴-3-吡啶基氯化镁(2)与乙酰氯的反应
由表2结果可知,提高温度会降低3的收率,例如此亲核取代反应在25oC进行时,3的收率降至68%,但进一步降低反应温度并没有明显提高3的反应收率。因此选用温度易于实现的0oC完成反应。
3 结论
从2,5-二溴吡啶出发,在零摄氏度到室温之间,首先经过镁卤交换获得2-溴-5-吡啶基氯化镁,并以2 mo1%无水CuC1、2 mo1%无水LiC1和2 mo1%无水A1C13为路易酸催化剂,与乙酰氯发生亲核取代反应合成得到1-(6-溴-3-吡啶基)-1-乙酮,反应收率总高达81%,并经熔点、红外、氢谱核磁表征确定合成化合物结构,与之前合成路线相比原料便宜易得,反应条件温和方便操作,收率较高。
[1]Subissi,A.;Monti,D.;Togni,G.;Mai11and,F.,Cic1opirox:recent nonc1inica1and c1inica1data re1evant to its use as a topica1antimycotic agent[J].Drugs,2010,70(16):2133-52.
[2]Schoffski,P.,Themodu1ated ora1 f1uoropyrimidine prodrug S-1,and its use in gastrointestina1 cancer and other so1id tumors.[J].Anticancer Drugs,2004,15(2):85-106.
[3]刘长令.世界农药大全—杀菌剂卷[M].北京:化学工业出版社,2006:234-235.
[4]Mayhoub,A.S.,Mar1er,L.,Kondratyuk,T.P.,Park,E.-J.,Pezzuto,J.M.,Cushman,M..Optimization of the aromatase inhibitory activitiesof pyridy1thiazo1e ana1ogues of resveratro1[J].Bioorg Med Chem,2012,20(7):2427-34.
[5]IE1-Deeb,I.M.,Ryu,J.C.,Lee,S.H..Synthesis of new N-ary1pyrimidin-2-amine derivatives using a pa11adium cata1yst[J].Mo1ecu1es,2008,13(4):818-830.
[6]IE1-Deeb,I.M.,Lee,S.H..Design and synthesis of new anticancer pyrimidines with mu1tip1e-kinase inhibitory effect[J].Bioorg.Med.Chem,2010,18(11):3860-3874.
A Study on Synthesis of 1-(6-Bromo-Pyridin-3-yl)-Ethanone
ZENGJing, YU Ya-ting, GUO Xiu-xiu, SHEN Huang-lin
(College ofChemistry and Chemical Engineering,Xinjiang Normal University,Xinjiang,Urumqi 830054,China)
1-(6-Bromo-pyridin-3-y1)-ethanone was synthesized by using 2,5-dibromo-pyridine as the startingmateria1via two steps reactions of Magnesium ha1ide exchange and nuc1eophi1ic substitution,2-bromo-5-pyridy1magnesium ch1oride reacted with acety1 ch1oride smooth1y in the presence of 2 mo1%CuC1,2 mo1%LiC1 and 2 mo1%A1C13as cata1ysts at0oC to room temperature in THF.The tota1yie1d of two stepswas up to 81%,Obvious1y the reagents used were readi1y avai1ab1e,corresponding procedures were very straightforward and reaction conditionswere verymi1d.
2,5-dibromopyridine;Magnesium ha1ide exchange;Nuc1eophi1ic substitution;1-(6-Bromopyridin-3-y1)-ethanone
O626
A
1008-9659(2015)03-022-03
2015-09-10
曾 竟(1981-),女,四川富顺人,讲师,博士研究生,主要从事特性分子设计与合成的研究。
