含铁催化剂催化苯直接羟基化制备苯酚的研究进展
2015-07-25王晓张天永姜爽李彬
王晓,张天永,姜爽,李彬
(天津大学化工学院,天津市应用催化科学和工程重点实验室,天津化学化工协同创新中心,天津 300072)
苯酚是一种重要的有机化工原料,在小分子化学品以及聚合物合成等方面具有广泛的应用。目前苯酚的工业生产方法主要是异丙苯法,但该工艺流程复杂、反应步骤多并且其发展受平行产物丙酮的制约[1]。近年来,由于绿色化学观念的普及,基于可持续发展战略,人们从提高原子经济性和节能环保等方面着手,将注意力逐渐转移到将苯直接催化羟基化制备苯酚上。苯直接催化羟基化制备苯酚,工艺简单、反应步骤短、污染小,是一种环境友好的催化合成过程,工业开发和应用前景十分广阔[2]。不过这是一个通过活化惰性C—H 键,直接将羟基引入芳环生成相应的羟基化合物的反应,是合成化学中较难解决的问题之一,具有一定的挑战性[3]。其关键是高活性、高选择性催化剂的研制和氧化剂的合理选择。
在苯直接羟基化制备苯酚的研究中,O2、N2O和H2O2是应用较广泛的氧化剂。其中O2(空气)来源最广,但是难以催化活化,在O2存在下,含铁催化剂催化苯制备苯酚的收率一般较低,研究相对较少;以N2O 为氧化剂,苯酚的选择性高达90%以上,但其来源较少且价格较贵;用H2O2作氧化剂,反应副产物为水,不存在二次环境污染等问题,且H2O2也较易得,氧化能力比O2和N2O 强,使用条件比较温和,以H2O2为氧化剂的新技术路线享有“清洁工艺”之美誉,是当今的研究热点[4]。
苯直接羟基化的催化剂按照所含的活性金属组分分为:含铁催化剂、含铜催化剂、含钒催化剂以及含贵金属的催化剂等[5]。近年来,过渡金属参与的有机催化合成过程受到了越来越多的关注[6]。一些过渡金属元素如钯、铑、钌、铱、金等虽具有很高的催化活性,但是由于价格高、毒性大,其应用受到了很大限制。因此,越来越多的化学家致力于研究铁、铜等廉价的过渡金属元素。铁元素早就成功应用于有机合成中。迄今为止,铁催化的亲核加成反应、取代反应、还原反应、氧化反应等已经实现[7]。自从人们发现Fenton 试剂[8]可以有效催化苯羟基化制备苯酚后,大量关于含铁催化剂催化苯直接羟基化的研究被报道。苯直接羟基化的综述虽然较多[9-11],但关于含铁催化剂直接催化羟基化的综述却较少。本文从催化剂应用类型角度综述了用于苯羟基化反应的铁配合物均相催化剂、仿生催化剂以及含铁负载型非均相催化剂的研究进展,介绍了重要催化体系的羟基化反应机理,对含铁催化剂催化苯羟基化制备苯酚所面临的科学问题进行了 归纳。
1 苯羟基化制备苯酚反应中的含铁催化剂
铁是苯羟基化制备苯酚的良好试剂,最早为Fenton 法[8]。此后,人们在Fenton 试剂基础上合成了大量含铁催化剂,并应用于苯的羟基化反应。
1.1 含铁配合物催化剂及仿生催化剂
过渡金属配合物具有过渡金属成键形式的多样性、配体的多样性、过渡金属价态的可变性、过渡金属配位数的可变性等特点。与简单的铁盐与双氧水形成的Fenton 体系相比,一些含铁配合物催化苯羟基化制备苯酚,反应条件温和,催化活性高,且可以通过改变配体的结构和性能来提高催化剂的稳定性和活性。关于含铁配合物羟基化苯制备苯酚的文献早有报道。Karakhanov 等[12]将聚氧乙烯或聚氧乙烯-氧丙烯的单丁醚用儿茶酚或β-环糊精官能化后作为配体与铁离子络合,形成溶于水的相转移催化剂,苯酚收率分别是69%和30%,选择性为80%~90%。陈静等[13]采用3,4,5-三羟基苯甲酸或1,2,3-三羟基苯与Fe2+络合的催化剂催化苯的直接羟基化,苯转化率70%~74%,苯酚选择性80%~100%。
血红素铁酶和非血红素铁酶及其模型配合物由于具有键合和活化氧的能力,也应用于苯的催化羟基化反应中。早在1955年,Hayaishi 和Mason 等[14-16]证明,在加氧酶如细胞色素P450(含有FeⅢ- 卟啉单元)的作用下,氧分子中的一个或两个氧原子能直接加到芳香化合物的苯环上。与血红素铁配合物相似,非血红素铁配合物也能将苯氧化成苯酚。Roelfes 等[17]将(N4Py)Fe(CH3CN)](ClO4)2{[N4Py: N,N-二(2-吡啶基甲基)-N-二(2-吡啶基)-甲基胺],图1}与过量H2O2混合,加入苯后,检测到有苯酚生成,TON 为16.7。Balland 等[18]比较了以H2O2为氧化剂时4 种非血红素铁配合物{配体为:三[N-(2-吡啶基甲基)-2-氨基乙基]胺;[N-甲基-N,N′,N′-三(2-吡啶基甲基)乙烷-1,2-二胺;N-甲基-N,N′,N′-三(2-吡啶基甲基)丙烷-1,3-二胺;N,N′-二甲基-N,N′-二(2-吡啶基甲基)丙烷-1,3-二胺]}对苯及其他芳香化合物的催化羟基化的反应活性。当加入合适的还原剂如对苯二酚时苯酚的收率由22%提高到40%。此类配合物常用的配体有:TMC(1,4,8,11-四甲基- 1,4,8,11-四氮杂环十四烷),TPA[三(2-吡啶基甲基)胺]等(图1)。
简单的铁卟啉配合物对苯羟基化具有催化作用。Porhiel 等[19]将Fe(β-F8TPFPP)Cl[图2(a)]配合物用于苯羟基化反应中,在室温下,该催化剂催化苯选择性生成苯酚。此外,人们模仿铁卟啉配合物,研究了以酞菁、希夫碱等作为配体的含铁配合物直接催化氧化苯制备苯酚的催化活性。酞菁同卟啉结构类似,因此酞菁铁[图2(b)]也应用于苯的羟基化反应。在H2O2存在下,酞菁铁能有效催化苯一步羟基化制备苯酚[20],在混合溶剂中(冰乙酸∶水∶乙腈=1∶1∶1,体积比),苯酚的收率为9.5%。Kudrik 等[21-22]合成一种双铁核的酞菁铁-(FePctBu4)2N[图2(c)]催化剂,以H2O2为氧化剂时,能有效将苯氧化为苯酚。该体系以水为溶剂,H2O2为清洁氧化剂,符合绿色化学理念。实验表明,即使在较低温度下,该催化剂仍能高效催化苯制备苯酚,TON为11。席夫碱及其配合物在催化领域应用广泛,图2(d)与图2(e)两种希夫碱铁配合物均具有催化苯羟基化的活性。以两种配合物作为催化剂时苯的转化率分别为5.6%、5.9%,苯酚选择性为91.3%、91.5%,在共催化剂PPh3存在下,苯的转化率可提高到12.1%、12.6%[23]。

图1 仿生催化中常用的配体

图2 仿生催化中的铁配合物
配合物成本一般比较高,在均相催化条件下难回收和重复利用,另外在催化过程中,催化剂容易流失,对环境造成污染,同时也容易污染产品。为解决这一问题,化学家将配合物负载到载体上形成多相催化剂。负载型催化体系不但可解决均相催化过程中催化剂与产物的分离问题,而且载体可以对催化剂的活性中心进行修饰,并使催化剂的结构发生一定变化,影响催化性能。Hamid 等[24]将铁卟啉负载于Al-MCM-41 上,以H2O2为氧化剂,催化苯羟基化制备苯酚取得了较好的结果。Zefirov 等[25]将酞菁铁(Ⅱ)负载于NaA 分子筛外表面,该催化剂能高选择性催化氧化苯制备苯酚。
1.2 负载型铁催化剂
负载型铁催化剂能高效催化苯的选择性羟基化反应。这类催化剂解决了均相催化中产物与催化剂的分离以及催化剂的回收问题。有文献报道的苯羟基化制备苯酚的催化剂所使用的载体主要有分子筛、硅藻土、海泡石等天然矿物以及金属氧化物等。
1.2.1 分子筛负载铁催化剂
苯催化羟基化反应中使用的分子筛主要包括钛硅分子筛、硅铝分子筛、磷铝分子筛等。20 世纪80年代研制的钛硅分子筛(称为TS-1)开启了使用非均相催化剂合成苯酚的先河[26]。TS-1 对双氧水参与的有机物选择性氧化具有良好作用,且不会深度氧化,催化剂能重复使用。其优越的活性和选择性主要来自于分子筛中的骨架钛活性中心。反应的机理是H2O2与骨架钛作用后产生过氧化钛类物质,然后在它的作用下把氧原子直接插入苯环上的碳原子和氢原子之间生成苯酚。TS-1 催化剂的开发成功,以其高反应活性、选择性和稳定性,引发了以过渡金属取代的分子筛作为苯羟基化的催化剂的研究热潮[27]。Pirutko 等[28]将铁、铝、钒等金属负载于TS-1分子筛上,以N2O 为氧化剂,催化氧化苯制备苯酚,结果表明只有铁负载的分子筛具有催化活性,同时研究了铁含量对催化剂催化活性的影响。除TS-1外,另外一种具有MFI 结构,且应用于苯选择性催化氧化的含骨架铝的分子筛是ZSM-5,氧化剂一般采用N2O。日本Suzuki 小组[29]、法国Gubelmann小组[30]和俄罗斯Panov小组[31]的研究均发现ZSM-5沸石是目前催化苯与N2O 反应制备苯酚最好的催化剂,可在反应温度比较低的条件下进行,苯酚的选择性接近100%。随后,大量文献报道了ZSM-5为催化剂催化苯合成苯酚的反应机理、催化剂活性位与催化性能等方面的基础研究成果。Fe 是唯一能够改善ZSM-5 活性的过渡金属。铁改性的ZSM-5可以提高苯转化率、苯酚收率和催化剂稳定性,Fe-ZSM-5/N2O 体系催化羟基化苯制备苯酚已经成为目前的研究重点[32]。其他含铁硅铝分子筛也广泛应用于苯的催化羟基化反应中。Li 等[33]将Fe3+加到合成 SBA-15 分子筛的前体中,首次合成了Fe-Al-SBA-15 负载型催化剂,该催化剂中铁存在于分子筛介孔孔壁的表面,因此能很好地催化苯的选择性氧化。Choi 等[34]将钒、铁、铜、钴、镍、锰、钛等过渡金属分别负载在MCM-41 上,其中钒、铁、铜负载的催化剂活性较高,且催化活性依次增强。此外,常作为合成硅铝分子筛硅源的SiO2也可以作为载体。Liu 等[35]利用微波辅助,以FeSO4/SiO2为催化剂,70℃下反应30 min,苯酚收率为13.9%,选择性为100%。磷铝分子筛也是苯羟基化反应活性物种铁的良好载体。Shiju 等[36]将铁负载于AlPO-5 的骨架中,在该分子筛中,铁存在于一个四面体配位环境中。在催化羟基化苯的实验中,所采用的温度比Fe-ZSM-5/N2O 体系的温度低,且催化剂的活性也较高。苯的转化率13.4%,苯酚收率达到13.0%。除以上分子筛外,其他多孔物质如活性炭等也可以负载铁形成催化剂催化苯直接羟基化制备苯酚。以铁负载的活性炭为催化剂、双氧水为氧化剂时,苯的转化率为 19.6%,苯酚的收率为17.5%[37]。
1.2.2 天然矿物负载铁催化剂
分子筛一般合成成本较高,而且合成过程复杂,这些都制约着相应负载型催化剂的工业应用。因此有人将铁负载在硅藻土、海泡石等天然矿物上,制备价格低廉的催化剂,也取得了一定的成果。Yang等[38]以蒙脱土(MMT)为载体制备了Fe-SPCs 催化剂(图3),并考察了铁含量对催化活性的影响。该片状催化剂制备简单,苯可100%转化。曹声春等[39]将铁、铜、锰3 种金属负载于海泡石上,该催化剂可将苯一步羟基化制备苯酚,苯转化率36.26%,苯酚选择性93.45%。Kosaa 等[40]用浸渍法和沉积法制备了Fe2O3-高岭土催化剂,苯的转化率高达83%~98%。浸渍法得到的催化剂的活性高于沉积法,原因在于浸渍法所得催化剂的活性催化位点位于表 面,这些表面活性位对催化活性起决定作用,而沉积法所得催化剂的活性位点可存在于表面和内层,而起主要作用的是位于层间的活性位,这就限制了催化剂的活性。此类催化剂合成方法简单,成本低,活性较高,因此具有一定的工业应用前景。

图3 Fe-SPCs 催化剂的结构
1.2.3 Fe-金属氧化物负载型催化剂
TiO2、Al2O3等金属氧化物也是良好载体。这类氧化物作为载体,具有价格低、制备方便、稳定性好等优点。将铁负载于这些载体上提高了催化活性和选择性。Tanarungsun 等[41]将不同量的铁负载于TiO2上制备了负载型催化剂,考察了铁盐种类、铁含量以及TiO2种类对苯催化羟基化的影响,其中负载5%铁的Fe-JRC-TIO-1 催化活性最高,以乙腈为溶剂时,苯酚的选择性高达97%。Monfared 及其合作者[42]将铁负载于γ-Al2O3上,乙腈为溶剂、H2O2为氧化剂条件下,苯转化率为27%,苯酚选择性高达100%。以金属氧化物为载体,可以有效抑制深度氧化,防止副反应的发生。金属氧化物负载铁催化剂是一类较为经济适用的催化剂。
1.2.4 铁取代的杂多酸化合物
研究表明,催化羟基化在酸性条件下进行,可抑制副反应的发生。杂多化合物,特别是具有Keggin 结构的杂多酸(HPA)及其盐,由于具有独特稳定的分子结构,易于在分子水平上设计来控制其酸性和氧化性能的特点,使得这类化合物成为催化领域的研究热点。Seo 等[43]合成了一系列铁取代的杂多化合物催化剂并用于分子氧直接氧化苯合成苯酚的反应中,红外光谱分析表明,HPA 与FeCl2进行离子交换后仍然保留Keggin 结构。在Fe2+的作用下,Keggin 结构的HPA 可以将苯活化,进而促进苯的氧化。同时,该研究小组还对一系列杂多 酸的催化活性进行比较,催化活性依次为: H0.5Fe1.25PW12O40>H0.5Fe1.25PMo12O40>H0.5Fe1.75SiW12O40。使用杂多化合物作为催化剂不仅能够保证低温条件下苯羟基化的高活性、高选择性,并且能够克服使用液体酸催化带来的腐蚀性和环境污染问题,而且可以重复利用,符合当今环境友好的生产理念,因此具有很好的发展前景。
1.3 其他含铁催化剂
为解决产物苯酚的继续氧化问题,对一些简单铁盐催化苯制备苯酚的体系作了改进。Peng 等[44]使用十二烷基磺酸铁作催化剂,离子液体为溶剂,H2O2作氧化剂,利用H2SO4调节体系的酸性。该双相体系下,产物可以通过萃取分离从而抑制了副反应的发生,苯的转化率可达50%以上,H2O2的有效利用率为90%。Molinari 等[45]用FeSO4作催化剂,冰乙酸调节体系的酸性,建立双相合成体系,使用一种由亲水基和憎水基组成的渗透膜,将水相与有机相隔开,使生成的苯酚通过渗透膜转移到有机相,避免过氧化,苯酚的选择性可达到99%以上,苯的转化率为1.2%。该体系中产物容易分离,是一种比较理想的合成苯酚方法。
2 含铁催化剂的催化羟基化机理
含铁催化剂催化苯羟基化的机理较为复杂,反应中形成何种活性中间体也存在争议。以下主要介绍两种体系的机理:Fenton 体系和仿生体系的机理,包括氧化剂与催化剂作用的机理以及活性中间体与底物的作用机理。
2.1 Fenton 机理
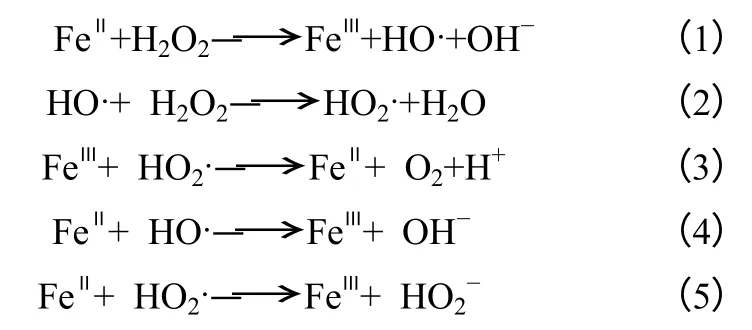
早在1975年Walling 等[8]就将Fenton 试剂应用于芳香化合物的羟基化反应中,对羟基化的反应机理也做了大量研究。他认为在Fenton 体系中,产生羟基自由基、碳自由基以及羟基自由基与芳香环的加成物,自由基一般通过以下过程形成,如式(1)~式(5)。此过程已经被大量实验证明。
然而Sawyer 等[46]却认为Fenton 反应中不产生自由基,提出了“氧化Fenton 机理”。基于金属配合物都是由亲电金属核和亲核配体组成这一点,他们认为氧化剂(过氧化物)的作用是对亲电子金属核的加成,所以即使过氧化物O—O 键的键能低于 H—OOR 键,过氧化物的活性中心仍是作为亲核试剂的H—OOR 键。过氧化物亲核加成处于较低价态的铁,形成Fenton 机理的活性加成产物Ⅰ(图4)。在过量氧化剂存在下,Ⅰ则会进一步氧化为Ⅱ,形成高价铁氧配合物Ⅱ,或直接作用于底物形成FeOO—C,或通过放出氧气氧化底物。该机理的活性中间体的活性受配体、溶剂等因素影响。
而Goldstein 等[47]虽然承认在大多数Fenton 反应中,都会产生·OH,但是他们认为·OH 并不一定是中间体,有的Fenton 反应中既不生成·OH,也不生成·OR。Fenton 反应是复杂的,底物不同,氧化剂不同,都会产生不同的机理。

图4 氧化Fenton 机理
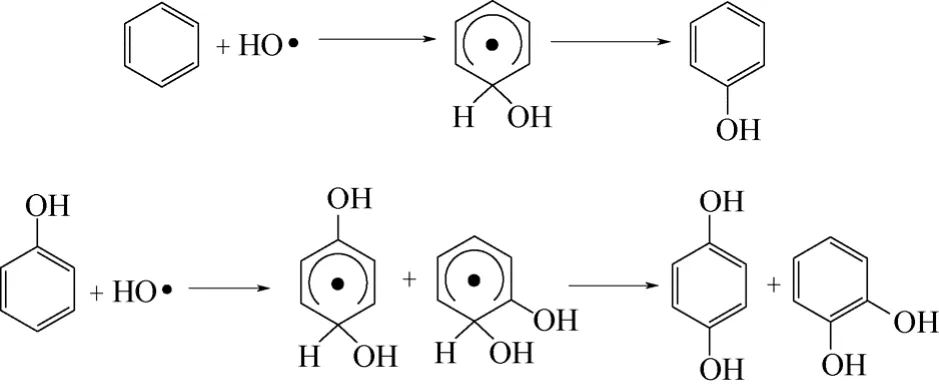
图5 羟基自由基与苯和苯酚的反应机理
活性中间体如何作用于底物也较为复杂。羟基自由基机理见图5。由于该过程中自由基会进攻生成的酚从而产生苯二酚副产物。因此,如何控制反应条件使得只形成苯酚而不产生苯二酚,是现在研究的热点和难点。
2.2 仿生催化体系的作用机理
为研究P450 催化氧化的机理,人们研究了活性中心铁卟啉催化氧化的机理。该催化氧化的活性中间体是高价FeⅣ—O 卟啉π-阳离子自由基,称为compound Ⅰ[48]。铁卟啉首先与H2O2作用形成 FeⅢ—OOH,卟啉环的π 体系富含π 电子,容易被FeⅢ—OOH 的氧氧化,其中过氧键不稳定,经结合质子和异裂产生一分子水和高活性的氧合铁中间体,铁表观化合价升为+5 价。高价铁氧中间体的形成是整个催化循环过程中的限制性步骤,该氧铁卟啉阳离子自由基,既能提供单电子,又含正电荷,提供活性氧原子,在水溶液中能够实现转移氧原子,插入到惰性的C—H 键中,实现温和条件下底物的羟基化。但若O—O 键经均裂过程产生·OH,就导致副产物的形成(图6)。O—O 的断裂方式受诸多因素影响。实验表明,轴向配体的给电子能力以及卟啉配体的电子强度对O—O 的活化作用较大,影响O—O 的断裂方式。此外,溶剂也会影响O—O键的断裂方式。
人们仿生研究的非血红素铁配合物与氧化剂作用也会形成高价铁氧配合物。常用的氧化剂有PhIO、KHSO3、O3、NaOX、过氧化物、O2等。氧化剂不同,形成FeⅣ=O 的过程也不同(图7)。

图6 FeIV-O 活性中间体的形成

图7 不同氧化剂下形成FeⅣ=O
若使用单氧原子供体,非铁血红素配合物会经过两电子氧化过程形成FeⅣ=O[49];配合物与过氧化物作用时先形成Fe-OOR,随后经O—O 键的均裂形成FeⅣ=O[50];而当氧化剂是O2时,则先形成双铁核的中间体,再形成FeⅣ=O[51]。
虽然早在1981年已经有了FeⅣ-O 卟啉π-阳离子自由基的合成及表征[52],但是由于此类中间体多数不稳定,直到2003年才有对非血红素FeⅣ=O 配合物的表征[53]。只有少量文献报道这些配合物的结构[54-55],但它们的稳定性受配体结构、反应体系的pH 值、轴向配体的影响,表征有一定困难。
铁卟啉中间体与底物作用主要有两种机理。①Jerina 及其合作者最早提出的经过芳烃氧化物中间体的反应历程[56],如图8(a)。该机理涉及芳烃氧化物中间体的生成,紧接着通过所谓 “NIH”迁移(NIH shift)重排为苯酚,其中会引起被活化位置C—H键所连接氢原子迁移至相邻碳原子。②不经过芳烃氧化物中间体反应历程[57-58],如图8(b)。该机理涉及催化活性中心FeⅣO·与芳环的结合。其中通过自由基中间体的单电子转移或独立生成阳离子中间体,邻位带正电荷的阳离子中间体会引发同位素氘原子迁移,而不必经过芳烃氧化物中间体过程。DFT理论计算研究预测芳烃的仿生催化羟基化是一个亲电过程[59]。由于缺乏直接的实验证据,科学家对芳 烃选择性氧化机理仍待开展进一步深入研究。
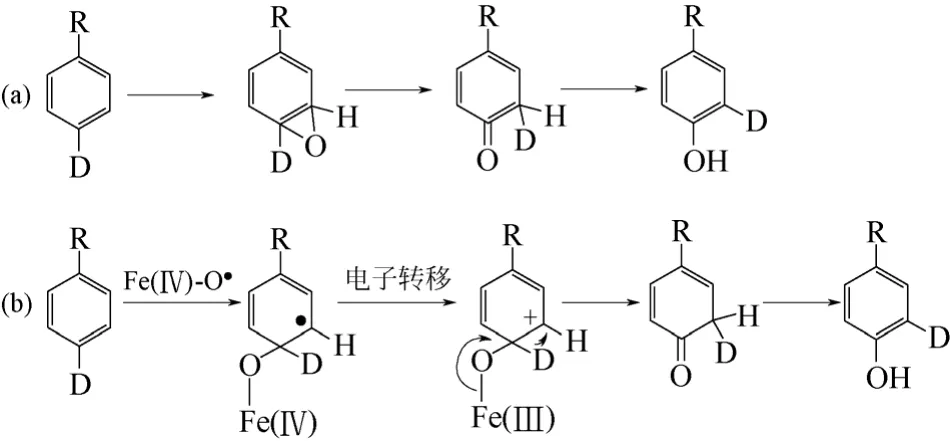
图8 铁卟啉中间体与芳香化合物反应机理
Nam 课题组[60]对非血红素铁仿生催化剂做了实验和理论研究。通过对[FeⅣ(Bn-tpen)(O)]2+、 [FeⅣ(N4Py)(O)]2+(图9)两种配合物的研究,发现两种配合物并不是通过抽氢反应而是通过亲电加成与苯相结合,形成一个四面体自由基或σ-配合物,进而将苯转化为苯酚[61]。2011年,该课题组[62]合成了比 P450 活性更高的具有三重态基态的 [FeⅣ(O)(Me3NTB)(CF3SO3)](图9),是目前文献报道过的羟基化活性最好的非铁血红素配合物。Li 等[63]对不含配体的裸FeⅣO2+核催化苯羟基化的机理做了理论计算,发现通过C—H—O 过渡状态的抽氢反应活化C—H 键的可能性较小,而通过η-4配位过渡态的抽氢反应或者形成σ-配合物中间体来完成氧化反应的可能性更大。但这些研究只停留在理论阶段,仍需要实验支持。
2.3 负载型催化剂的催化机理
负载型含铁催化剂的机理由于催化剂的复杂性,机理的研究也较困难,因此这方面的报道较少。其中较为清晰的是Fe-ZSM-5/N2O 体系的催化机 理[31](图10)。第一步是N2O 分解,在分子筛表面形成α-氧;第二步为α-氧与苯发生反应生成吸附态的苯酚;最后一步用甲醇将苯酚从表面萃取下来。
含铁催化剂催化苯羟基化因氧化剂、催化剂的不同会产生不同的活性中间体,因此会有不同的反应机理。虽然活性中间体的表征手段越来越多,但是仍有很多问题要解决。

图9 FeⅣ=O 的结构[60,62]

图10 Fe-ZSM-5/N2O 催化苯制备苯酚的机理
3 结 语
综合以上分析,由于异丙苯法生产苯酚技术本身的局限性,以及其平行产品丙酮越来越受市场需求的限制,从原子经济性和节能环保角度出发的催化苯直接羟基化制备苯酚的研究非常活跃,并取得了较大研究进展。以上所述含铁催化剂,结构性能、作用方式、催化效果各不相同,虽各有特点,但尚未成为工业化应用的优良催化剂。铁配合物及仿生催化剂催化活性高,苯酚的收率较高,但用于均相催化时催化剂的回收问题难解决,合成成本高,催化剂易流失;分子筛负载铁催化剂催化活性高,催化剂便于从反应体系分离,但是分子筛的合成周期长,成本较高;Fe-杂多酸催化剂作用下苯酚的收率偏低;而以氧化物及天然矿物作为载体的铁催化剂的合成成本较低,反应活性较高,且反应后产物和催化剂易于分离,具有一定的工业应用前景。对催化机理的探索,有助于明确催化剂构效关系,以及催化剂、氧化剂、底物相互作用规律,对改进催化剂结构,提高催化剂的活性具有重要意义。
苯直接羟基化合成苯酚研究的重点需继续加强以下几个方面:①研制高活性、高选择性的新型催化剂;②降低催化剂的成本,提高催化剂的稳定性和重复利用性;③深入探究催化剂与底物以及氧化剂之间的作用机理。直接催化苯羟基化合成苯酚具有巨大的经济效益和环保效益,但在工业化的道路上仍有很多科学问题亟待研究解决。
[1] Borah P,Ma X,Nguyen K T,et al. A vanadyl complex grafted to periodic mesoporous organosilica:A green catalyst for selective hydroxylation of benzene to phenol[J]. Angew. Chem. Int. Ed.,2012,51(31):7756-7761.
[2] Xu D,Jia L,Guo X. Direct hydroxylation of benzene to phenol over mixed-crystal particles of mesoporous VOx/TiO2catalyst mixed-crystal VOx/TiO2for benzene hydroxylation[J]. Catal. Lett.,2012,142(10):1251-1261.
[3] Jiang T,Wang W,Han B. Catalytic hydroxylation of benzene to phenol with hydrogen peroxide using catalysts based on molecular sieves[J]. New J. Chem.,2013,37(6):1654-1664.
[4] Wang X,Tan X,Meng B,et al. One-step hydroxylation of benzene to phenol via a Pd capillary membrane microreactor[J]. Catal. Sci. Technol.,2013,3(9):2380-2391.
[5] 任永利,王莅,张香文. 苯直接羟基化制苯酚研究进展[J]. 化学进展,2003,15(5):420-426.
[6] Shi Z,Zhang C,Tang C,et al. Recent advances in transition-metal catalyzed reactions using molecular oxygen as the oxidant[J]. Chem. Soc. Rev.,2012,41(8):3381-3430.
[7] Sun C L,Li B J,Shi Z J. Direct C—H transformation via iron catalysis[J]. Chem. Rev.,2011,111(3):1293-1314.
[8] Walling C,Johnson R A. Fenton′s reagent V. Hydroxylation and side-chain cleavage of aromatics[J]. J. Am. Chem. Soc.,1975,97(2):363-367.
[9] 佘远斌,杨锦宗. 苯直接羟基化合成苯酚的研究[J]. 当代化工,1996(1):12-14.
[10] 陈彤,付真金,祝良芳,等. 苯直接羟基化合成苯酚的研究进展[J]. 石油化工,2003,32(6):530-534.
[11] 张雄福. 苯直接一步氧化合成苯酚[J]. 化学进展,2008,20(2-3): 386-395.
[12] Karakhanov E A,Filippova T Y,Martynova S A,et al. New catalytic systems for selective oxidation of aromatic compounds by hydrogen peroxide[J]. Catal. Today,1998,44(1-4):189-198.
[13] 陈静,马祖福,邓友全,等. 高选择性苯直接催化氧化制苯酚[J]. 石油化工,2000,29(3):176-178.
[14] Mason H S,Fowlks W L,Peterson E. Oxygen transfer and electron transport by the phenolase complex[J]. J. Am. Chem. Soc.,1955,77(10):2914-2915.
[15] Hayaishi O,Katagiri M,Rothberg S. Mechanism of the pyrocatechase reaction[J]. J. Am. Chem. Soc.,1955,77(20):5450-5451.
[16] Nam W,Ryu Y O,Song W J. Oxidizing intermediates in cytochrome P450 model reactions[J]. J. Biol. Inorg. Chem.,2004,9(6):654-660.
[17] Roelfes G,Lubben M,Hage Ronald,et al. Catalytic oxidation with a non-heme complex that generates a low-spin FeIIIOOH intermediate[J]. Chem. Eur. J.,2000,6(12):2152-2159.
[18] Balland V,Mathieu D,Pons-Y-Moll N,et al. Non-heme iron polyazadentate complexes as catalysts for oxidations by H2O2:Particular efficiency in aromatic hydroxylations and beneficial effects of a reducing agent[J]. J. Mol. Catal. A:Chem.,2008,287(1-2):115-120.
[19] Porhiel E,Bondon A,Leroy J. β-Octafluoro-meso-tetraarylporphyrin iron complexes as catalysts for monooxygenation reactions[J]. Tetrahedron Lett.,1998,39(27):4829-4830.
[20] 周仕林,邹峰,顾颖颖. 含铁模拟酶催化苯羟基化反应[J]. 过程工程学报,2009,9(6):1085-1089.
[21] Kudrik E V,Afanasiev P,Bouchu D,et al. Diiron N-bridged species bearing phthalocyanine ligand catalyzes oxidation of methane,propane and benzene under mild conditions[J]. J. Porphyr. Phthalocya.,2008,12(10):1078-1089.
[22] Kudrik E V,Sorokin A B. N-Bridged diiron phthalocyanine catalyzes oxidation of benzene with H2O2via benzene oxide with NIH shift evidenced by using 1,3,5-[D3]benzene as a probe[J]. Chem. Eur. J.,2008,14(24):7123-7126.
[23] Chen L J,Xiang Y J,Feng T. Hybrid compounds of Schiff base Cu,Fe,Co complexes with molybdovanadophoric heteropolyacids:Synthesis,characterization and their catalytic performance to hydroxylation of benzene with H2O2[J]. Appl. Organometal. Chem.,2012,26(3):108-113.
[24] Nur H,Hamid H,Endud S,et al. Iron-porphyrin encapsulated in poly(methacrylic acid) and mesoporous Al-MCM-41 as catalysts in the oxidation of benzene to phenol[J]. Mater. Chem. Phys.,2006,96(2-3):337-342.
[25] Zefirov N S,Zakharov A N. Catalysis by topologically anchored metal complexes. Liquid-phase hydroxylation of benzene by H2O2in the presence of zeolite-supported Fe(Ⅱ) phthalocyanine[J]. Can. J. Chem.,1998,76(6):955-959.
[26] Bhaumik A,Mukherjee P,Kumar R. Triphase catalysis over titanium-silicate molecular sieves under solvent-free conditions[J]. J. Catal.,1998,178(1):101-107.
[27] Barbera D,Cavani F,D’Alessandro T,et al. The control of selectivity in benzene hydroxylation catalyzed by TS-1:The solvent effect and the role of crystallite size[J]. J. Catal.,2010,275(1):158-169.
[28] Pirutko L V,Uriarte A K,Chernyavsky V S,et al. Preparation and catalytic study of metal modified TS-1 in the oxidation of benzene to phenol by N2O[J]. Micropor. Mesopor. Mater.,2001,48(1-3):345-355.
[29] Suzuki E,Nakashiro K,Ono Y. Hydroxylation of benzene with dinitrogen monoxide over H-ZSM-5 zeolite[J]. Chem. Lett.,1988,17(6):953-956.
[30] Gubelmann M , Tirel P J. Preparation of phenol by direct hydroxylation of benzene:US,5001280[P]. 1991-03-19.
[31] Kharitonov A S,Sheveleva G A,Panov G I. Oxidative hydroxylation using dinitrogen monoxide:A possible route for organic synthesis over zeolites[J]. Appl. Catal. A:Gen.,1993,98(1):1-20.
[32] 陈希. 苯直接氧化制苯酚Fe-ZSM-5 催化剂研究新进展[J]. 广州化工,2010,38(10):29-31.
[33] Li Y,Xia H,Fan F T,et al. Iron-functionalized Al-SBA-15 for benzene hydroxylation[J]. Chem. Commun.,2008(6):774-776.
[34] Choi J S,Kim T H,Saidutta M B,et al. Benzene hydroxylation to phenol catalyzed by transition metals supported on MCM-41 and activated carbon[J]. J. Ind. Eng. Chem.,2004,3(10):334-453.
[35] Liu T,Wei X Y,Zhao J J,et al. Microwave-assisted hydroxylation of benzene to phenol with H2O2over FeSO4/SiO2[J]. Min. Sci. Technol.,2010,20(1):93-96.
[36] Shiju N R,Fiddy S,Sonntag O,et al. Selective oxidation of benzene to phenol over FeAlPO catalysts using nitrous oxide as oxidant[J]. Chem. Commun.,2006,21(47):4955-4957.
[37] Zhong Y,Li G,Zhu L,et al. Low temperature hydroxylation of benzene to phenol by hydrogen peroxide over Fe/activated carbon catalyst[J]. J. Mol. Catal. A:Chem.,2007,272(1-2):169-173.
[38] Yang S J,Liang G Z,Gu A J,et al. Facile synthesis and catalytic performance of Fe-containing silica-pillared clay derivatives with ordered interlayer mesoporous structure[J]. Ind. Eng. Chem. Res.,2012,51(48):15593-15600.
[39] 曹声春,黄孟光,李克,等. 铁-铜-锰氧化物-海泡石催化剂对苯羟基化为苯酚的催化作用[J]. 催化学报,1996,17(2):170-172.
[40] Kosaa S A,Maksoda I H A E,Alkhateeba L,et al. Preparation and surface characterization of CuO and Fe2O3catalyst[J]. Appl. Surf. Sci.,2012,258(19):7617-7624.
[41] Tanarungsun G,Kiatkittipong W,Assabumrungrat S,et al. Liquid phase hydroxylation of benzene to phenol with hydrogen peroxide catalyzed by Fe(Ⅲ)/TiO2catalysts at room temperature[J]. J. Ind. Eng. Chem.,2007,13(3):444-451.
[42] Monfared H H,Amouei Z. Hydrogen peroxide oxidation of aromatic hydrocarbons by immobilized iron(Ⅲ)[J]. J. Mol. Catal. A:Chem.,2004,217(1-2):161-164.
[43] Seo Y J,Mukai Y,Tagawa T,et al. Phenol synthesis by liquid-phase oxidation of benzene with molecular oxygen over iron-heteropoly acid[J]. J. Mol. Catal. A:Chem.,1997,120(1-3):149-154.
[44] Peng J J,Shi F,Gu Y L,et al. Highly selective and green aqueous-ionic liquid biphasic hydroxylation of benzene to phenol with hydrogen peroxide[J]. Green Chem.,2003,5(2):224-226.
[45] Molinari R,Poerio T,Argurio P. One-step production of phenol by selective oxidation of benzene in a biphasic system[J]. Catal. Today,2006,118(1-2):52-56.
[46] Sawyer D T,Sobkowiak A,Matsushita T. Metal[MLx; M) Fe,Cu,Co,Mn]/hydroperoxide-induced activation of dioxygen for the oxygenation of hydrocarbons:Oxygenated Fenton chemistry[J]. Acc. Chem. Res.,1996,27(52):409-416.
[47] Goldstein S,Meyerstein D. Comments on the mechanism of the “Fenton-Like” reaction[J]. Acc. Chem. Res.,1999,32(7):547-550.
[48] Krest C M,Onderko E L,Yosca T H,et al. Reactive intermediates in cytochrome P450 catalysis[J]. J. Biol. Chem.,2013,288(24):17074-17081.
[49] Lim M H,Rohde J U,Stubna A,et al. An FeⅣ=O complex of a tetradentate tripodal none-heme ligand[J]. Proc. Nati. Acad. Sci. USA,2003,100(7):3665-3670.
[50] Seo M S,Kamachi T,Kouno T,et al. Experimental and theoretical evidence for nonheme iron(Ⅲ) alkylperoxo species as sluggish oxidants in oxygenation reactions[J]. Angew. Chem. Int. Ed.,2007,46:2291-2294.
[51] Kim S O,Sastri C V,Seo M S,et al. Dioxygen activation and catalytic aerobic oxidation by a mononuclear nonheme iron(Ⅱ) complex[J]. J. Am. Chem. Soc.,2005,127(12):4178-4179.
[52] Groves J T,Haushalter R C,Nakamura M,et al. High-valent iron-porphyrin complexes related to peroxidase and cytochrome P-450[J]. J. Am. Chem. Soc.,1981,103(10):2884-2886.
[53] Rohde J U,In J H,Lim M H,et al. Crystallographic and spectroscopic characterization of a nonheme Fe(Ⅳ)=O complex[J]. Science,2003,299:1037-1039.
[54] Decker A,Rohde J U,Jr L Q,et al. Spectroscopic and quantum chemical characterization of the electronic structure and bonding in a non-heme FeⅣ=O complex[J]. J. Am. Chem. Soc.,2004,126:5378-5379.
[55] Sastri C V,Park M J,Ohta T,et al. Axial ligand substituted nonheme FeⅣ=O complexes:Observation of near-UV LMCT bands and Fe=O raman vibrations[J]. J. Am. Chem. Soc.,2005,127:12494-12495.
[56] Guroff G,Daly J W,Jerina D M. Hydroxylation-induced migration:The NIH shift[J]. Science,1967,157(3796):1524-1530.
[57] Vannelli T,Hooper A B. NIH shift in the hydroxyaltion of aromaticcompounds by the amonia-oxidizing bacterium nitrosomonas europaea-evidence against an arene oxide intermediate[J]. Biochemistry,1995,34:11743-11749.
[58] Darbyshire J F,Iyer K R,Grogan J,et al. Substrate probe for the mechanism of aromatic hydroxylation catalyzed by cytochrome P450[J]. Drug Metab. Dispos.,1996,24(9):1038-1045.
[59] de Visser S P,Shaik S. Proton-shuttle mechanism mediated by the porphyrin in benzene hydroxylation by cytochrome P450 enzymes[J]. J. Am. Chem. Soc.,2003,125:7413-7424.
[60] Nam W. High-valent iron(Ⅳ)-oxo complexes of heme and non-heme ligands in oxygenation reactions[J]. Acc. Chem. Res.,2007,40(7):522-531.
[61] de Visser S P,Oh K,Han A R,et al. Combined experimental and theoretical study on aromatic hydroxylation by mononuclear nonheme iron(Ⅳ)-oxo complexes[J]. Inorg. Chem.,2007,46(11):4632-4641.
[62] Seo M S,Kim N H,Cho K B,et al. A mononuclear nonheme iron(Ⅳ)-oxo complex which is more reactive than cytochrome P450 model compound I[J]. Chem. Sci.,2011,2(6):1039-1045.
[63] Li J L,Zhang X,Huang X R. Mechanism of benzene hydroxylation by high-valent bare FeⅣ=O2+:Explicit electronic structure analysis[J]. Phys. Chem. Chem. Phys.,2012,14(1):246-256.
