分子模拟技术在炼油领域的应用
2015-06-24周涵,任强,龙军
周 涵, 任 强, 龙 军
(中国石化 石油化工科学研究院, 北京 100083)
分子模拟技术在炼油领域的应用
周 涵, 任 强, 龙 军
(中国石化 石油化工科学研究院, 北京 100083)
简要介绍了近几年来分子模拟技术在炼油领域的应用,如各化学法炼制过程反应化学研究、渣油结构特征的研究、油品添加剂分子设计以及炼油催化剂的开发等。分子模拟作为一种能模拟炼油过程细节的有效工具已经在炼油工业各个领域的研究中发挥了重大作用。
分子模拟;反应化学;油品添加剂;催化剂
近年来,随着化学科学和计算机科学发展与提高以及不同计算化学方法取得的进展,分子模拟技术作为人们对过程细节进行科学研究的一项新的有效工具,得到越来越广泛的应用。分子模拟技术在炼油领域,如对各炼制过程核心转化规律的认识、渣油团聚物结构研究、油品添加剂分子设计以及分子筛催化剂等方面的应用,可以帮助研究人员更深入地认识理解所研究的反应体系,有利于从分子水平上提炼关键科学问题,揭示技术关键,催生技术创新思路,以便选择更合理的研发途径,更快地进行催化剂的改性和开发以及油品添加剂新产品的研制。分子模拟技术的应用对于进一步推动石油化工的科研创新,也具有十分重要的意义。
1 分子模拟技术在炼油领域的应用
1.1 反应化学方面研究
在炼油领域,石油的转化涉及大量的化学反应,其反应网络复杂,反应路径多变,反应机理多样。随着分子模拟在炼油反应化学规则方面的应用,从分子水平上进行了深入的认识和发现,大量的反应机理被揭示出来,为炼油技术的研发和改进提供了较大的帮助。
1.1.1 炼油反应中的化学反应规律认识
炼油过程中的化学反应复杂,种类繁多。通常,都是依靠实验结果推测炼油中的各种化学反应规律,而借助分子模拟技术,则可以从基元反应入手,从本质上认识反应化学规律。龙军等[1]利用分子模拟方法,研究了异丁烷-丁烯烷基化反应体系中C4和C8正碳离子可能进行的各类基元反应。结果表明,对于C4正碳离子而言,2种伯丁基正碳离子的能量明显较高,而仲丁基正碳离子和叔丁基正碳离子的能量分别比伯丁基正碳离子低71.94 kJ/mol和125.99 kJ/mol;对于C8正碳离子而言,所有的伯正碳离子在结构优化时,均自发地异构成了相应的C8仲正碳离子或叔正碳离子,说明异丁烷-丁烯烷基化反应体系中C4和C8伯正碳离子存在的概率很低,并依据量子化学计算结果,确定了异丁烷-丁烯烷基化反应体系中可能存在的正碳离子中间体,构建了具有较强预测能力的基元反应网络。鲁玉莹等[2]采用MS QMERA中QM和MM(量子力学和分子力学)相结合的方法,选用84T的HY分子筛簇模型,研究了HY分子筛催化C4烷基化反应中异丁烯质子化反应过程。结果表明,异丁烯首先吸附在分子筛上形成π-络合物,再通过正碳离子的过渡态生成表面烷氧基团;质子化反应的活化能垒比单纯H+进攻异丁烯的能垒高,此研究表明固体酸催化烷基化反应中正碳离子的引发要引入空间效应因素。
于宁等[3-4]采用基于密度泛函理论的量子化学方法研究了催化重整过程中正庚烷脱氢生成烯烃的反应过程。结果表明,在Pt催化剂作用下,烷烃脱氢反应能垒被有效地降低了。同时,对催化重整过程中2-庚烯生成正碳离子及正碳离子的移位和环化过程研究表明,在L酸活性中心作用下, 2-庚烯催化重整的主要产物具有五元环结构。该研究结果从理论角度首次论证了Mills催化重整反应机理的合理性,并进一步丰富与发展了催化重整反应机理,对于新型重整催化剂和工艺的开发具有重要意义。
康承琳等[5]采用分子模拟方法计算了二甲苯的分子轨道、3种二甲苯与H+之间的相互作用,以及二甲苯异构化过程中2种分子内反应机理的能量路径和过渡态。结果表明,苯环骨架异构(简称1,3-迁移)的过渡态路径比甲基在苯环上转移(简称1,2-迁移)路径复杂,且1,3-迁移路径过渡态最高能垒值220.5 kJ/mol远高于1,2-迁移的过渡态最高能垒85.1 kJ/mol。由此得出甲基在苯环上转移的1,2-迁移为二甲苯分子内异构化反应的主要机理路径,为1,3-迁移和1,2-迁移两种长期争论的分子内机理给出了清晰的证据,更深入地认识了二甲苯异构化反应的本质。
陶海桥等[6]利用密度泛函理论计算了正十六烷分子链中C4—H键和与之相邻的C3—C4键和C4—C5键质子化形成的C4HH、C3HC4和C4HC5五配位正碳离子的结构和能量。结果表明CHH五配位正碳离子能够转化为CHC五配位正碳离子。刘俊等[7-8]利用量子化学密度泛函的方法,研究了烃分子中C—C键键长变化规律,重点研究了吸电子取代基、给电子取代基对烃分子中C—C键键长分布的影响。同时研究了自由基机理和正碳离子机理这2种反应中间产物的结构与特性,从而解释这两种反应都容易发生β断裂的原因,并对这两种反应的条件进行了比较,这些研究为裂化反应复杂的反应机理提供了进一步的认识。
对于稠环芳烃的加氢特点也已有大量的研究工作。Duan等[9]以含有3个芳环渺位缩合结构的稠环芳烃分子作为模型化合物,采用量子化学理论,通过Hyperchem模拟软件对模型化合物的键长、键级进行计算比较。结果表明,环烷烃、脂肪链相比芳香环结构更容易首先发生加氢、断键反应;通过前线轨道理论的计算表明,稠环芳烃极易在贵金属催化剂表面发生吸附,并最易在稠环芳烃的γ碳上首先加氢,加氢反应的能垒为559.82 kJ/mol,反应热为-736.38 kJ/mol。刘锋[10]等对芘在不同温度下、不同催化剂催化的加氢反应进行了实验对比研究。张成等[11]采用量子力学方法,研究了氢自由基与多环芳烃的基元反应,结果表明,芘倾向于在端环边位先加氢,有3条主要的加氢路径;在反应初期,中间环边位碳原子的加氢稍占优势,但随着反应的深入进行,继续加氢变得困难;不同温度下芘在NiMo催化剂上加氢得到的反应产物分布的模拟计算结果与实验数据吻合较好。王春璐等[12]构建了一系列不同芳环数的稠环芳烃,以分子模拟为手段从反应机理层面研究稠环芳烃(PAHs)加氢反应过程;采用基于密度泛函理论的DMol3模块对反应过程进行模拟计算,得到不同稠环芳烃加氢过程的反应热和能垒,探索稠环芳烃按照自由基热反应机理加氢时反应的难易程度。结果表明,在自由基足量存在且能与模型化合物有效接触的理想状态下,稠环芳烃分子的芳环数、分子饱和度对其加氢难易程度影响不大;芳环极易与氢自由基结合,重质油加氢困难并非是因为稠环芳烃与氢自由基直接反应困难造成。这些工作为多环芳烃的加氢提供了新的研究方法。
对于供氢剂的供氢机理研究工作也取得了一些新的认识。宗士猛等[13]采用基于密度泛函理论计算了不同类型1-烷基四氢萘分子中α位C—H键断裂的反应能垒和反应热,讨论了1-烷基四氢萘的供氢能力。研究表明,1-叔丁基四氢萘中的叔丁基破坏了α位C—H键断裂产物自由基的p-π共轭结构,导致1-叔丁基四氢萘的供氢能力低于四氢萘。杨哲等[14]以四氢萘为模型化合物,利用基于密度泛函理论的量子化学从头计算方法,对四氢萘的几何结构和电子结构进行了系统研究,得到了四氢萘不同位置C—H键和C—C键的键长、键级、键能以及电子云密度、前线轨道分布等微观结构信息,有助于加深对氢化芳烃反应特性的认识与理解。
1.1.2 脱硫、脱氮反应机理
分子模拟技术较为广泛的应用于炼油工业中脱硫、脱氮这一领域,为脱硫、脱氮技术的改进提供了有利的技术支持。
龙军等[15]采用量子化学理论计算方法,对S Zorb脱硫反应机理进行了深入研究。结果表明,S Zorb技术的工艺过程实质上是高选择性催化加氢超深度脱硫过程,而不是简单的吸附过程。在S Zorb技术中,通过在加氢催化剂中添加H2S吸收组分ZnO,可有效地转移加氢脱硫过程中产生的H2S,建立一个H2S分压极低的反应环境,避免H2S与汽油中高辛烷值烯烃组分生成硫醇的副反应,同时使催化剂活性金属Ni处于零价态而具有对噻吩类含硫化合物很高的吸附活性,但对高辛烷值烯烃、芳烃组分仅有很低的吸附活性。在此基础上,提出了催化加氢-H2S吸收转移协同作用的催化加氢吸附脱硫机理,并指出保持催化剂中Ni处于零价态避免生成NiS是提高催化加氢脱硫选择性的关键。工业应用结果表明,S Zorb 技术在实现超深度脱硫的同时具有很好的辛烷值保留能力。该研究为指导工业催化剂的研制提供了重要的理论支持。
Moses等[16-17]分别计算了噻吩在MoS2和CoMoS催化剂上Mo边和S边的加氢脱硫(HYD)和直接脱硫(DDS)的反应路径的能量变化,如图1所示。对于MoS2催化剂,噻吩比较容易在Mo边的Brim位上吸附并加氢,而在S边的S空位上C—S键断裂容易。与之前的结论不同,该研究认为,在配位饱和的Mo边(Brim位,无S空位),噻吩也可以加氢生成2,5-二氢噻吩,并继续发生S—C断键;而在S边,反应可按HYD路径和DDS路径进行,S边本身虽不具有活性,但产生S空位后拥有反应活性,S空位的生成能垒与H2分压有关。对于CoMoS催化剂,认为Co的加入降低了HYD途径和活性中心再生的能垒,增加了C—S键断裂的能垒。李皓光等[18]应用密度函数计算方法研究了H2在超深度加氢脱硫催化剂体系(MoS,CoMoS,NiMoS,NiWS,WS,CoWS)上吸附解离的化学过程,考察了在特定催化剂活性相结构模型的不同位置上H2解离的催化活性。结果表明,催化剂不同反应位上的催化活性不同,边位的催化活性较高。H2在几种不同加氢脱硫催化剂上的反应热及能垒计算结果显示了它们对H2吸附解离反应的催化活性不同,在NiMoS和NiWS催化剂上,边位的氢解活性高,而MoS、CoMoS催化剂上(001)层面S原子的氢解活性高。还比较了助剂在H2解离活化反应中的间接、直接作用,发现其间接作用大于直接作用。
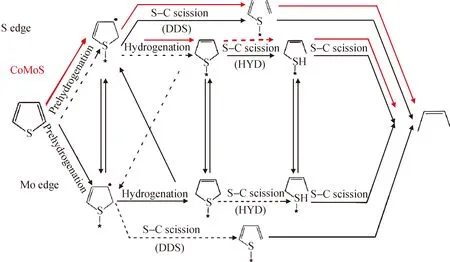
图1 噻吩加氢脱硫反应路径
李倩等[19]从烟气中NOx在吸附剂表面形成硝酸盐的事实出发,结合理论计算研究了硝酸盐被还原的微观反应过程。以H2为还原剂、Al(NO3)3为模型化合物,采用密度泛函方法计算得到的结果表明,H2还原Al(NO3)3的反应分步进行。H2吸收热量,解离成H·,H·将Al(NO3)3还原为Al(OH)3和NO,然后NO进入气相与H2反应生成N2。H2解离成H·的反应是整个反应的控速步骤,除H2离解形成H·反应之外的其余步骤均为放热反应。这些研究为脱硫、脱氮技术提供了新的研究思路。
1.2 重油结构的模拟研究
重油的结构组成及其聚集状态一直是研究人员关注的问题和难题之一。由于实验条件的限制,还无法通过实验方法获得重油的真实结构组成。采用分子模拟方法,针对重质油这一领域对体系分子的尺寸、黏度、聚集等物理形态等方面进行模拟计算,为重油的研究开阔了思路。
任强等[20]采用耗散粒子动力学方法,从介观尺度描述重油的胶体结构,并研究了重油稳定性与其胶体结构之间的关系。结果表明,重油体系以胶体的形式存在,胶粒是由一些从内到外极性逐渐变小的组分组成,在胶体体系中作为分散相,而饱和分和单环芳烃为胶体体系的分散介质;重油体系的活化状态受温度的影响较大,并存在一个最佳活化温度;重油体系随着剪切速率的增加,体系的剪切黏度降低,表现出假塑性流体的特性。该结果与实验结果一致,但该研究方法无法给出沥青质分子间的聚集体微观结构。
Roberto等[21]使用半经验量子力学方法AMI,研究实验获得的和由量子力学方法计算所得的烷基苯IR谱。基于对这些谱图的研究,分析2927 cm-1和2957 cm-1峰强度比值与烷基苯碳链的n(CH2)/n(CH3)之间的线性关系,确定了3种巴西减压沥青中胶质、沥青质中的烷基侧链的相对分子质量。任文坡等[22]结合核磁共振表征技术与分子动力学手段,对重油组分的密度和结构进行了模拟验证。潘月秋等[23]以几种稠环芳烃作为模型化合物,利用量子化学中的密度泛函理论进行结构优化模拟,对稠环芳烃分子尺寸的计算为分子筛的开发提供了指导。高媛媛[24]利用溶剂萃取和色谱手段从不同来源的重质油中分离出多种卟啉化合物,并采用紫外-可见光谱和质谱手段分析了几类卟啉化合物的结构,以其中5种具有代表性的卟啉化合物作为模型化合物,模拟计算了它们的结构、极性,从而预测了其从重油中分离的难易程度。
1.3 在油品添加剂中的应用
分子模拟技术可以帮助研究人员分析和了解油品添加剂的作用机理及复合效应,为深入研究添加剂的结构和性能之间的关系提供直观、高效、可靠的信息,在辅助油品添加剂的设计及新型添加剂的开发方面具有较好的应用前景。
刘琼等[25-26]用分子模拟研究了摩擦改进剂烷基链特性对减摩性能的影响。通过对摩擦改进剂作用机理分析,提出利用描述分子内旋转难易程度的分子柔顺性来定量反映摩擦改进剂烷基链结构对减摩性能的影响。采用HFRR测定含不同碳链结构脂肪酸摩擦改进剂油品的摩擦系数,利用Kier柔顺性计算方法计算不同烷基链脂肪酸摩擦改进剂的分子柔顺性,然后将二者进行关联。结果表明,油品摩擦系数与摩擦改进剂分子柔顺性有较好的关联性, 摩擦改进剂分子柔顺性可作为定量反映其烷基链对减摩性能影响的特性参数。
长期以来,人们大多是采用传统的实验方法研究屏蔽酚抗氧剂的作用机理和结构性能关系。而近年来,借助于计算机分子模拟技术,人们也获得了许多新的认识。Pfaendtner等[27]采用B3LYP泛函方法对小分子屏蔽酚和ROO·相互作用的10个反应进行了量子化学计算,发现在所有反应中均存在氢键中间体,但是氢键中间体的存在不影响反应的平衡、反应活化能ΔEa和反应焓变ΔHrxn。苏朔等[28-29]结合实验方法和量子力学方法,研究了含硫屏蔽酚(SHP)的分子结构与其抗氧性能之间的关系。采用压力差示扫描量热法(PDSC)测定了6种SHP(A、B、C、D、E、F)的氧化诱导期(OIT),应用广义梯度近似的密度泛函理论(DFT-GGA)分析了它们的前线分子轨道性质,并计算了O—H键的解离能BDE(O—H)以及H、O和S原子的Mulliken净电荷布局数。结果表明, A、B、C、D和E 4种SHP分子中,酚羟基和硫醚2种抗氧官能团均可以同时发挥抗氧活性,而F则是酚羟基首先发挥抗氧活性;BDE(O—H)和S原子的Mulliken净电荷数与OIT均有较强的相关性,BDE(O—H)与S原子的Mulliken净电荷数相关性较强。
在低温流动改进剂研究方面,李妍等[30]采用实验和分子动力学模拟结合的方法,考察了多种乙烯-醋酸乙烯酯(EVA)和聚甲基丙烯酸酯(PMA)类低温流动改进剂(CFI)在蜡晶形成阶段对蜡晶晶形的影响。结果表明,在蜡晶形成阶段,CFI通过改变柴油烃分子的排列方式和相互作用能,控制蜡晶形态,避免粒径较大且容易聚集的片状蜡晶生成。CFI分子保持伸展的构象,并对烃分子有较强的吸附能力,更利于其实现控制晶形的作用。CFI分子中的极性基团和非极性短支链可起到分散烃分子和保持聚合物分子的伸展构象的作用。CFI聚合物分子中含有较多连续的亚甲基结构有利于增强其吸附烃分子的能力,给合成新的低温流动改进剂提供了有利的理论依据。
1.4 炼油催化剂方面的应用
分子模拟技术作为炼油催化剂研究与开发的重要辅助工具,在炼油催化剂领域的各个方面,如分子筛的辅助合成设计、吸附与扩散等方面均得到了广泛的应用,可以为科研工作者节省大量繁琐的探索性实验。
1.4.1 新型催化剂的辅助设计
分子模拟技术是催化剂尤其是分子筛催化剂的辅助设计开发的一种有效工具。研究人员可通过分子模拟得到催化过程的反应机理,从而确定研发催化剂的思路。
梁晓青等[31]采用密度泛函理论计算方法,对碱金属Li、Na、K、Rb、Cs改性的SAPO-34分子筛的L酸结构进行了理论研究,得出碱金属改性分子筛中Li—O键长最短,从能量上看,Li改性分子筛的稳定性最好。另外,计算了CO和NH3在碱金属Li、Na、K、Rb、Cs改性分子筛L酸上的吸附情况,发现Li改性分子筛的吸附能量最大,对分子筛结构影响最大, L酸酸性最强,其他碱金属改性分子筛的L酸酸性依次减弱。李金芝等[32]以密度泛函理论(DFT)为基础,对丁烯-1骨架异构单分子机理反应过程模拟研究后发现,丁烯-1很难在新鲜分子筛催化剂的酸性位上通过单分子反应生成异丁烯,而是以分子筛催化剂酸性位上无法化学脱附的正碳离子为新的活性位,进行第2次反应,生成异丁烯。因为该种反应方式可以使速控步骤的能垒大幅降低,有利于提高异丁烯的选择性。因此而提出的有效利用第1次反应的吸附物种,引发后续烯烃骨架异构化反应的结论,对新型分子筛催化剂的设计提供了思路。张宝吉等[33]利用分子力学和分子动力学计算工具计算了Na+的各种水合结构及能量,以此研究水合Na+所起的结构生成剂的作用。用Monte Carlo Docking方法对各种结构的水合Na+在MFI分子筛微孔结构内的附着行为进行了模拟计算,结果表明,配位数为偶数时水合阳离子的结构较为合理。这种合理性体现在,当配位数为偶数时,Na+-nH2O 体系中金属离子的位置与氧原子的位置呈现对称结构,Na+-4H2O 和Na+-6H2O是相对比较合适的结构,在MFI结构分子筛的合成中起模板剂作用的是Na+-4H2O结构。其计算结果可为研究分子筛合成中其他水合离子的结构生成和结构破坏作用提供借鉴。
张丽伟等[34]对P、Fe改性的ZSM-5分子筛粉末进行X射线扫描,获得了较高分辨率的衍射数据,并由此得到分子筛的化学组成。采用分子模拟软件中的建模及结构优化工具,构建了硅/铝比为23.77的ZSM-5分子筛的三维空间构型,用分子力学方法计算Fe2+在分子筛孔道中的位置,并用Rietveld精修的方法验证Fe2+在ZSM-5分子筛孔道中的位置。结果表明,Fe改性的ZSM-5分子筛中Fe2+主要存在于十元环S形孔道和直孔道交叉处,少量存在于直孔道中。计算了Fe2+在孔道内的配位间距,对改性的ZSM-5分子筛进行了结构参数分析,最终确定了2种Fe2+在孔道内的确切坐标分数,为催化剂的研发提供了较多的基础数据。
1.4.2 催化剂的吸附和扩散性能
催化剂的吸附和扩散性质对确定催化剂所能达到的选择性十分重要。以前由于缺乏进行预测的理论根据,每一个有研究价值的体系的扩散系数必须通过实验测定。分子模拟技术的发展及应用,为研究催化剂的吸附和扩散性质、温度对扩散系数的影响、选择合适的催化剂提供了很好的工具,诸多研究人员在这方面做了大量的研究工作[35-36]。
(1) 气体在分子筛中扩散系数的分子模拟
扩散系数可反映分子筛孔道骨架结构与吸附质的相互作用,以及吸附质分子在分子筛晶体内微孔中的扩散速率[37]。
Krishna等[38-39]计算了纯组分甲烷、乙烷和丙烷的 M-S 扩散系数,并研究了 M-S 扩散系数、自扩散系数和交换系数之间的关系;还计算了不同孔道结构分子筛的 M-S 扩散系数和交换系数,分析发现,交换系数与 M-S 扩散系数、负载量通过一常数相关联,而常数的大小表示了分子在分子筛孔道内的受限制程度。Jobic等[40]对比了实验方法和分子动力学方法测得的CF4在MFI型分子筛中的 M-S 扩散系数,结果一致。由以上可见,分子动力学方法可以合理计算分子的扩散系数,用以描述一些扩散行为。
Zimmermann等[41]发现,对于同一模拟体系,晶胞的刚性和柔性状态对于扩散系数的影响非常复杂,但他们没有给出确切的规律性的结果。Demontis等[42]还认为,以时间为函数计算扩散系数或大的客体分子在分子筛内扩散时,将分子筛骨架设定为刚性状态是不合理的,希望能通过更高级的动力学模拟软件和并行计算机出现来解决目前存在的问题。
(2) 吸附质负载量和温度对扩散系数的影响
吸附质负载量是分子筛扩散性质重要的影响因素。一般说来,随着吸附质负载量的增加,吸附质分子之间的作用力增强,扩散行为受到限制,扩散系数降低。Rungsirisakun等[43]应用分子动力学方法研究了在FAU 分子筛中分别放置 2、4、6、8 个苯分子时苯的自扩散系数,发现苯的扩散系数随着其分子个数的增加而下降。Krishna等[44]采用分子动力学方法研究了7种吸附质(He,Ne,Ar,Kr,H2,N2,CO2)在全硅分子筛上的扩散系数,发现在一定范围内随着负载量的增加,扩散系数先降低后增加到最大值再逐渐降低。由于考察范围和体系的差异,以上研究出现了不同的结果,也没有合理的解释。Dubbeldama等[45]在研究中发现同样的现象,甲烷和乙烷在 LTA型分子筛中的扩散系数也是先降低后增加到最大值再逐渐降低,并结合自由能的变化解释了这种现象。他们认为,随着负载量的增加,分子与分子间的作用力不断增强,限制了分子的扩散,扩散系数也随之降低;负载量再增加,分子之间的排斥力克服了分子与分子筛的吸引力,使得自由能垒降低了,扩散系数出现了最大值;随着负载量的继续不断增加,分子之间的作用力占了主导地位,扩散系数又不断降低。Nanok等[46]研究甲醛在NaX分子筛上扩散随负载量变化时也出现相同的规律,并用集体效应(Collective effects)和位阻效应(Steric hindrance)解释了出现这一规律的原因。
同样,温度对扩散系数的影响也可以通过分子模拟来研究。Goktug等[47]采用分子动力学方法研究了不同温度下MTBE在 silicalite-1 中的扩散系数,得到负载量一定的情况下,随着温度的升高扩散系数增加的结果。Plant等[48]研究了甲烷在NaY分子筛中不同温度下的扩散系数。结果表明,随着温度的提高,甲烷的扩散系数不断增加。
袁帅等[49]应用分子模拟技术计算了不同芳烃和环烷烃分子最低能量构象的分子尺寸(a×b×c),并计算了其在MFI、FAU分子筛中的扩散能垒。计算结果表明,分子的最小截面尺寸(a×b)与扩散能垒相关。苯、萘、环戊烷、环己烷分子可以在MFI分子筛中扩散,其它多环芳烃、多环环烷烃分子均很难扩散。分子在MFI直孔道中扩散比在正弦孔道中容易。FAU分子筛由于孔径尺寸较大,分子在其孔道内扩散比在MFI孔道中容易,而两超笼间的十二元环会限制芘、全氢芘及比之更大的分子在FAU孔道中扩散。从计算结果推断催化裂化原料中的重油大分子只有先在分子筛或其他活性材料表面一次裂化成一定尺寸的小分子,才能扩散进入MFI、FAU分子筛晶内发生进一步反应,这与实验观察到的结果是一致的。
(3) 在金属催化剂及金属氧化物催化剂上的吸附
除了分子筛外,金属及金属氧化物也是常用的炼油催化剂,对于这类催化剂的研究,应用分子模拟技术,也取得了较好的结果。姚淑娟等[50]采用密度泛函理论(DFT)中广义梯度近似方法(GGA),研究了Pt原子与γ-Al2O3的(001) 面的相互作用。计算结果表明,Pt团簇能够稳定吸附在该表面,Pt原子在表面O位的吸附能明显较高,这主要是由Pt向基底O原子转移了电子所致;电荷分析表明,Pt原子显正电性,Pt和Al原子之间存在排斥作用;计算的平均吸附能大小依赖于Pt团簇的大小和形状,总体趋势是随着Pt原子数增多,吸附能降低。郭大为[51]采用分子动力学MD方法、周期边界性的从头算法研究了γ-Al2O3的表面和本体,及与H2S、H2O、MoS2等小分子的反应性能。利用分子力学研究了γ-Al2O3的晶体结构及(100)面和(110)面的结构数据,发现SO2在(100)面的吸附能力要强于其在(110)面的能力,NO在(110)面的吸附能力大于SO2,并在吸附实验中得到与理论预测相同的结论,为催化剂的制备提供了新的思路。
2 结束语
随着计算机技术的发展和模拟方法的不断更新,分子模拟技术已经可以对石油这种复杂体系及石油炼制这种复杂过程的研究提供越来越有效的帮助,由此也使炼油科研人员从分子水平思考炼油技术创新成为可能。迄今为止,分子模拟的应用范围已经扩展到炼油领域的各个环节,涉及的体系越来越复杂,研究的尺度越来越多样,研究的问题越来越深入,对所研究的炼油过程的技术进步提供了分子水平上的、更深入的认识,甚至是新的知识。可以预见,随着研究工作的不断拓展、不断深入,分子模拟在炼油技术的创新中将发挥越来越重要的作用。
[1] 龙军, 何奕工, 代振宇. 异丁烷-丁烯烷基化体系基元反应网络研究[J].石油学报(石油加工), 2010, 26(1): 1-7.(LONG Jun, HE Yigong, DAI Zhenyu. Elucidation of elementary reaction network for isobutane-butene alkylation system [J]. Acta Petrolei Sinica(Petroleum Processing Section), 2010, 26 (1): 1-7.)
[2] 鲁玉莹,李永祥, 龙军,等. HY催化C4烷基化中异丁烯质子化反应的分子模拟[J]. 石油学报(石油加工), 2014, 30(5):765-771.( LU Yuying, LI Yongxiang,LONG Jun,et al. Molecular simulation of isobutene protonation in C4 alkylation catalyzed by HY[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2014, 30(5):765-771.)
[3] 于宁, 龙军, 周涵, 等. 正庚烷脱氢生成烯烃反应的分子模拟[J]. 石油学报(石油加工), 2013, 29(2):181-185.(YU Ning, LONG Jun, ZHOU Han, et al. Molecule simulation of dehydrogenation of n-heptane to produce olefins[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2013, 29(2):181-185.)
[4] 于宁, 龙军, 马爱增, 等. 2-庚烯碳正离子移位及环化反应的分子模拟[J]. 石油学报(石油加工), 2013, 29(4):549-554.(YU Ning, LONG Jun, MA Aizeng, et al. Molecule simulation of transpositions and cyclization reactions for 2-heptene carbonium[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2013, 29(4):549-554.)
[5] 康承琳,龙军, 周震寰,等. 二甲苯异构化的反应化学[J]. 石油学报(石油加工), 2012, 28(4):533-537.(KANG Chenglin,LONG Jun,ZHOU Zhenhuan,et al. Reaction chemistry of xylene isomerization[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2012, 28(4): 533-537.)
[6] 陶海桥,龙军,周涵,等. 正十六烷CHH和CHC五配位正碳离子之间转化反应的分子模拟[J]. 计算机与应用化学, 2011, 28(3):347-350.(TAO Haiqiao,LONG Jun,ZHOU Han, et al. Molecular simulation studies on isomerization of CHH and CHC carboniumions of protonatedn-hexadecane[J]. Computers and Applied Chemistry, 2011, 28(3):347-350. )
[7] LIU Jun,LONG Jun, HE Zhenfu, et al. Research on C—C bond length distribution in hydrocarbon molecules[J]. China Petroleum Processing and Petrochemical Technology, 2010, 12 (3): 6-13.
[8] 刘俊,龙军,贺振富. 自由基和正碳离子结构方面的异同[J]. 石油学报(石油加工), 2011, 27 (2): 168-174. (LIU Jun,LONG Jun,HE Zhenfu, et al. Similarities and differences between the structures of free radical and carbenium ion[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2011, 27 (2): 168-174.)
[9] DUAN A J, XU C M, GAO J S, et al. Molecular simulation for catalytic hydrotreatment of coker heavy gas oil derived from Athabasca bitumen [J]. Journal of Molecular Structure, 2005, 734 (1-3): 89-97.
[10] 刘锋, 李会峰, 刘泽龙, 等. 芘在不同加氢催化剂上反应性能的比较[J]. 石油学报(石油加工), 2011, 27(3):343-347.(LIU Feng, LI Huifeng, LIU Zelong, et al. Comparison of pyrene hydrogenation over different catalysts[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2011, 27(3): 343-347.)
[11] 张成, 周涵, 王丽新, 等. 芘加氢的分子模拟研究[J]. 计算机与应用化学, 2012, 29(2): 161-164.(ZHANG Cheng, ZHOU Han, WANG Lixin, et al. Molecular simulation on hydrogenation of pyrene[J]. Computers and Applied Chemistry, 2012, 29(2):161-164. )
[12] 王春璐, 周涵, 王子军, 等. 稠环芳烃直接加氢的分子模拟研究[J]. 计算机与应用化学, 2012, 29(10):1221-1224.(WANG Chunlu, ZHOU Han, WANG Zijun, et al. Molecular modelling of direct radical hydrogenation on different PAHs molecules [J]. Computers and Applied Chemistry, 2012, 29(10):1221-1224.)
[13] 宗士猛,龙军,周涵, 等. 烷基取代基对烷基四氢萘供氢能力影响的分子模拟[J]. 石油学报(石油加工), 2012, 28(5): 705-710.( ZONG Shimeng,LONG Jun,ZHOU Han, et al. Molecular simulation of the impact of alkyl substituent on hydrogen donating ability of alkyl tetralin[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2012, 28 (5): 705-710.)
[14] 杨哲,宗士猛,龙军. 四氢萘微观结构的量子化学研究[J]. 计算机与应用化学, 2012, 29(4): 465-468.(YANG Zhe,ZONG Shimeng,LONG Jun. Quantum mechanical studies on the micro-structure of tetrahydro-naphthalene[J]. Computers and Applied Chemistry, 2012, 29(4): 465-468.)
[15] 龙军,林伟,代振宇. 从反应化学原理到工业应用Ⅰ S Zorb技术特点及优势[J]. 石油学报(石油加工), 2015, 31(1):1-6.(LONG Jun,LIN Wei,DAI Zhenyu. From detailed desulfurization mechanism to successful commercial application ⅠFeatures and advantages of S Zorb technology[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2015, 31(1): 1-6.)
[16] MOSES P G, HINNEMANN B, TOPS∅E H, et al. The hydrogenation and direct desulfurization reaction pathway in thiophene hydrodesulfurization over MoS2catalysts at realistic conditions: A density functional study[J]. Journal of Catalysis, 2007, 248(2):188-203.
[17] MOSES P G, HINNEMANN B, TOPS∅E H, et al. The effect of Co-promotion on MoS2catalysts for hydrodesulfurization of thiophene: A density functional study[J]. Journal of Catalysis, 2009, 268(2):201-208.
[18] 李皓光, 赵晓光, 周涵, 等. 氢分子在超深度加氢脱硫催化剂上吸附与解离的量子力学研究[J]. 石油学报(石油加工). 2007, 23(1):51-57.(LI Haoguang, ZHAO Xiaoguang, ZHOU Han, et al. Quantum-mechanical calculation of chemisorption and activation of hydrogen on ultra-deep hydrodesulfurization catalysts[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2007, 23(1):51-57.)
[19] 李倩, 郭大为, 周涵. 氢气还原硝酸盐的理论研究[J]. 石油学报(石油加工), 2013, 29(3):427-432.(LI Qian, GUO Dawei, ZHOU Han. Theoretical study on hydrogen reduction of nitrate[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2013, 29(3):427-432.)
[20] 任强, 代振宇, 周涵. 重油胶体结构的介观模拟[J]. 石油学报(石油加工), 2013, 29(1): 86-94.(REN Qiang, DAI Zhenyu, ZHOU Han. Mesoscale simulation on colloidal structures of heavy oil[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2013, 29 (1): 86-94.)
[21] COELHO R R, HOVELL I, MARISA B, et al. Characterization of aliphatic chains in vacuum residues (VRs) of asphaltenes and resins using molecular modelling and FTIR techniques [J]. Fuel Processing Technology, 2006, 87(4): 325-333.
[22] 任文坡, 陈宏刚, 杨朝合. 重油组分密度表征和结构验证的分子模拟[J]. 化工学报, 2009, 60(8): 1883-1888.(REN Wenpo, CHEN Honggang, YANG Chaohe, et al. Applicat ion of molecular simulat ion in density characterization and structure validation of heavy oil fractions [J]. Journal of the Chemical Industry and Engineering Society of China, 2009, 60(8): 1883-1888.)
[23] 潘月喜, 王大喜, 高金森. 重油特征分子尺寸的精确计算方法[J]. 石油学报(石油加工), 2007, 23(4): 63-67. (PAN Yueqiu, WANG Daxi, GAO Jinsen. The accurate calculating method for molecular dimensions of heavy oil character molecules [J]. Acta Petrolei Sinica(Petroleum Processing Section, 2007, 23 (4): 63-67.)
[24] 高媛媛. 两种原油卟啉化合物结构特征及其对重油催化裂化影响研究[D〗. 上海: 华东理工大学, 2011.
[25] 刘琼, 龙军, 武志强, 等. 摩擦改进剂烷基链特性对减摩性能的影响[J]. 石油学报(石油加工), 2014, 30(2): 189-193.(LIU Qiong, LONG Jun, WU Zhiqiang, et al. Effect of alkyl chain characteristic of friction modifier on friction-reducing[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2014, 30(2): 189-193.)
[26] 刘琼, 龙军, 武志强, 等. 摩擦改进剂烷基链特性的研究[C〗// 全国第十七届润滑脂技术交流会论文集, 广西,2013:40-43.
[27] PFAENDTNER J, BROADBELT L J. Elucidation of structure-reactivity relationships in hindered phenols via quantum chemistry and transition state theory[J]. Chemical Engineering Science, 2007, 62(18-20): 5232-5239.
[28] 苏朔, 龙军, 段庆华, 等. 含硫屏蔽酚抗氧剂构效关系的密度泛函理论研究[C〗// 第五届国际分子模拟与信息技术应用学术会议论文集, 武汉,2010: 736-746.
[29] 苏朔. 润滑油基础油的化学组成与其粘温性能的关系[J]. 原油及加工科技信息, 2009, (3):110-129.(SU Shuo. The relation between chemical composition and viscosity-temperature properties of lube base oil [J].Scientific and Technical Information of Petroleum Processing, 2009, (3):110-129.)
[30] 李妍, 赵毅, 段庆华, 等. 低温流动改进剂在柴油蜡晶形成阶段的晶形控制作用[J]. 计算机与应用化学, 2014, 31(4): 431-437.(LI Yan, ZHAO Yi, DUAN Qinghua, et al. Controlling crystal morphology mechanism of cold flow improver in formation stage of diesel wax crystal[J]. Computers and Applied Chemistry, 2014, 31(4): 431-437.)
[31] 梁晓青, 康丽华. 改性对SAPO-34分子筛L酸性影响的理论计算研究[J]. 石河子大学学报(自然科学版), 2012, (05):639-642.(LIANG Xiaoqing, KANG Lihua. Effect of modification on the L acid of SAPO-34 molecular sieve[J]. Journal of Shihezi University(Natural Science), 2012, (05):639-642.)
[32] 李金芝, 龙军, 赵毅, 等. 丁烯-1骨架异构反应机理的分子模拟研究[J]. 石油学报(石油加工), 2013, 29(2):7-11.(LI Jinzhi, LONG Jun, ZHAO Yi, et al. A density functional study of the mechanism of skeletal isomerization of butane-1[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2013, 29(2):7-11.)
[33] 张宝吉, 任强. 钠离子在分子筛合成中的结构导向作用的分子模拟研究[J]. 计算机与应用化学, 2009, 26(5):677-681.(ZHANG Baoji, REN Qiang. Molecular simulation studies on the structure directing effects of hydrate Na cation in the synthesis of microPorous materials[J]. Computers and Applied Chemistry, 2009, 26(5):677-681.)
[34] 张丽伟, 周涵, 代振宇, 等. 铁离子在ZSM-5分子筛中存在位置的理论研究[J]. 计算机与应用化学, 2012, 29(6):684-686.(ZHANG Liwei, ZHOU Han, DAI Zhenyu, et al. Predicting locations of non-framework Fe cations in zeolite ZSM-5[J]. Computers and Applied Chemistry, 2012, 29(6):684-686.)
[35] MANGA E D, BLASCO H,DA-COSTA P,et al. Effect of gas adsorption on acoustic wave propagation in MFI zeolite membrane materials: Experiment and molecular simulation[J]. Langmuir, 2014, 30(34):10336-10343.
[36] GANESH H S, PUNNATHANAM S N. Effect of adsorbate loading on selectivity during adsorption of C14/C15 and C15/C16n-alkane binary mixtures in silicalite[J]. Adsorption, 2014, 20(5-6), 729-736.
[37] 刘立凤, 赵亮, 陈玉, 等. 分子在分子筛上扩散行为的分子模拟研究进展[J]. 化工进展, 2011, 30(7):1406-1415.(LIU Lifeng, ZHAO Liang, CHEN Yu, et al. Research progress of molecular simulation of diffusion in zeolites[J]. Chemical Industry and Engineering Progress, 2011, 30(7):1406-1415.)
[38] KRISHNA R, VANBATEN J M. An investigation of the characteristics of Maxwell-Stefan diffusivities of binary mixtures in silica nanopores [J]. Chemical Engineering Science, 2009, 64(5):870-882.
[39] KRISHNA R, VANBATEN J M. Unified Maxwell-Stefan description ofbinary mixture diffusion in micro- and meso- porous materials[J].Chemical Engineering Science, 2009, 64(13):3159-3178.
[40] JOBIC H, SKOULIDAS A I, SHOLL D S. Determination of concentration dependent transport diffusivity of CF4 in silicalite by neutron scattering experiments and molecular dynamics[J]. Journal of Physical Chemistry B, 2004, 108(30):10613-10616.
[41] ZIMMERMANN N E R, JAKOBTORWEIHEN S, BEERDSEN E, et al. In-depth study of the influence of host-framework flexibility on the diffusion of small gas molecules in one-dimensional zeolitic pore systems[J]. Journal of Physical Chemistry C, 2007, 111(46):17370-17381.
[42] DEMONTIS P, SUFFRITTI G B. A comment on the flexibility of framework in molecular dynamics simulations of zeolites[J]. Microporous and Mesoporous Materials, 2009, 125(1-2):160-168.
[43] RUNGSIRISAKUN R, NANOK T, PROBST M, et al. Adsorption and diffusion of benzene in the nanoporous catalysts FAU, ZSM-5 and MCM-22: A molecular dynamics study[J]. Graphics Modell, 2006, 24(5):373-382.
[44] KRISHNA R, VANBATEN J M. Insights into diffusion of gases in zeolites gained from molecular dynamics simulations[J]. Microporous Mesoporous Materials, 2008, 109(1-3):91-108.
[45] DUBBELDAMA D, BEERDSEN E, VLUGT T J H, et al. Molecular simulation of loading-dependent diffusion in nanoporous materials using extended dynamically corrected transition state theory[J]. The Journal of Chemical Physics, 2005, 122(22):224712-224717.
[46] NANOK T, VASENKOV S, KEIL F J, et al. Molecular dynamics simulation study of the concentration dependence of the self-diffusivity of methanol in NaX V zeolite[J]. Microporous and Mesoporous Materials, 2010, 127(3):176-181.
[47] GOKTUG A M, KARVAN O, ERDEM-SENATALAR A. MTBE adsorption and diffusion in silicalite-1[J]. Microporous Mesoporous Materials, 2008, 115(1-2):93-97.
[48] PLANT D F, MAURIN G, BELL R G. Diffusion of methanol in zeolite NaY: A molecular dynamics study[J]. Journal of Physical Chemistry B, 2007, 111(11):2836-2844.
[49] 袁帅,龙军,周涵, 等. 芳烃、环烷烃分子在MFI和FAU分子筛中扩散行为的分子模拟[J]. 石油学报(石油加工), 2011, 27(4):508-515.(YUAN Shuai,LONG Jun,ZHOU Han, et al. Molecular simulation for the diffusion characteristics of aromatic and naphthenic hydrocarbons in the channel of MFI and FAU zeolites[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2011, 27(4):508-515.)
[50] 姚淑娟, 邵鑫, 崔守鑫, 等. Pt原子在γ-Al2O3(001)表面的吸附及迁移[J]. 物理化学学报, 2011, 27(8):1816-1822.(YAO Shujuan, SHAO Xin, CUI Shouxin, et al. Adsorption and migration of Pt atoms onγ-Al2O3(001) surface[J]. Acta Physico-Chimica Sinica, 2011, 27(8):1816-1822.)
[51] 郭大为.γ-Al2O3表面吸附SO2、NOx的机理分析[J]. 石油学报(石油加工), 2010, 26(2):235-241.(GUO Dawei. Analysis of adsorption mechanism of SO2 and NOxon surfaces ofγ-Al2O3[J]. Acta Petrolei Sinica(Petroleum Processing Section), 2010, 26(2):235-241.)
Applications of Molecular Simulation Technology in the Field of Oil Refining
ZHOU Han, REN Qiang, LONG Jun
(ResearchInstituteofPetroleumProcessing,SINOPEC,Beijing100083,China)
A brief review on the applications of molecular simulation technology in the field of oil refining, such as chemical reaction rules for all kinds of chemical refining process, the structure of residue, molecular design of oil additives and development of refining catalysis in recent years was carried out. Molecular simulation as an effectual implement which can investigate details of refining process has played an important role in the fields of refining industry.
molecular simulation; chemical reaction rule; oil additives; catalysts
2014-10-31
周涵,男,教授级高级工程师,博士,从事分子模拟和计算化学方面研究;Tel:010-82368717; E-mail:zhouh.ripp@sinopec.com
1001-8719(2015)02-0360-09
TQ028; O6
A
10.3969/j.issn.1001-8719.2015.02.016
