极锋以南洋区N2O源汇特性初析
2015-01-27詹力扬陈立奇张介霞李玉红吴曼许苏清
詹力扬 陈立奇 张介霞, 李玉红 吴曼 许苏清
(1国家海洋局第三海洋研究所,海洋大气化学与全球变化重点实验室,福建厦门361005;2厦门大学海洋与地球学院,福建厦门361005)
0 引言
N2O温室气体对全球气候和大气化学过程均有重要影响。等浓度N2O的温室效应是CO2的200—300倍。同时,其光化学产物NO在平流层中可与O3反应,破坏大气臭氧层[1]。在人类限制生产使用氟氯烃并致其大气浓度逐年下降的情况下,N2O成为21世纪排放量最大的臭氧层破坏气体[2]。因此,N2O相关研究受到日益关注。海洋是大气N2O最主要的来源之一。20世纪90年代之前,大多数研究[3-10]显示,海洋对大气 N2O储库的贡献量在10 Tg·a-1以内。在此基础上,IPCC[11]估算全球N2O来源为17.7 Tg·a-1,其中,约有3 Tg·a-1源于海洋。Nevison和Suntharalingam等[12-13]分别运用模型对全球海洋N2O源强进行评估,得到4 TgN·a-1的基本一致的结论。
在全球的主要洋区中,南大洋(本研究对南大洋定义是40°S以南的洋区)是全球海洋中十分重要的一个洋区,其面积约占世界大洋总面积的20%。它连接全球三大洋,除起到化学和生物物质传输通道作用外,它与其他大洋交换作用也强烈影响全球气候。而南大洋对全球变化效应具有放大作用,通常可以作为全球变化的进程指示器,因此南大洋成为研究全球变化的重要区域。此外,南大洋还是许多水团,如模态水、中间水、深层水和底层水团形成的区域。水团在南大洋的下沉过程为人为排放的CO2等温室气体提供了向大洋内部输送的通路。研究结果显示,30°S以南洋区吸收人为排放CO2体积约为全球大洋吸收量的1/3—1/2[14]。由此可见,南大洋在全球温室效应的调控方面起着极为重要的作用。
遗憾的是,时至今日,由于南大洋和南极海域的极端环境和恶劣天气成为地球大洋最难以接近和到达的海域,因此在南大洋进行过的N2O调查研究工作十分有限,但是这些有限的结果还是令人感兴趣的。Priscu等[15]对Ross冰架水体(Ross Ice Shelf或RIS)水体NH4+、N2O的研究工作,其结果显示RIS水体中N2O和大气基本持平,即非大气N2O的源区也非汇区。Weiss等[16]对南印度洋进行调查发现近南极某些区域表层海水具有较高的N2O饱和度;Rees等[17]对南大洋开展N2O研究中发现,德雷克海峡表层海水N2O分压与大气平衡;别林斯高晋海表层海水在季节融冰水稀释的作用下,略呈不饱和状态。根据这一研究结果,Rees等[17]认为,Bouwman等[18]提出南极海洋(Antarctic Ocean)为全球大气的重要来源的结论可能需要重新审视。Law等[19]在SOIREE南大洋施铁肥试验中(49°S—61°S纬度范围)调查了水体中N2O分布,结果显示调查区域表层海水N2O分压和大气平衡,在施放铁肥后观测到80 m处N2O浓度显著升高。Zhan和Chen[20]对南大洋印度洋和普里兹湾表层海水进行调查,提出物理因素为南大洋表层海水N2O分布主控因素。
模型研发和运用突破了现场条件限制。根据Nevison[12]利用现场调查共60 000个数据估算得到全球大洋的表层海水大气ΔpN2O,并利用美国国家大气研究中心(NCAR)获得的气象数据估算海气传输系数(gas transfer coefficiency),最后得到4 TgN·a-1的大气海洋年通量数据(范围1.2—6.8 TgN·a-1之间)。南大洋可能是全球N2O的一个重要来源,占全球海洋来源的35%;Suntharalingam等[13]运用海洋综合环流模型(ocean general circulation model,OGCM),结合海洋中ΔN2O和AOU关系随深度的变化,对N2O在海水中的垂直分布进行模拟,得到与Nevison[12]相类似的结果,得出全球海气通量值3.85 TgN·a-1(范围在2.7—8.0 TgN·a-1),其中,南大洋的释放通量占总通量的43%。在他们的研究中,均提出南大洋是全球N2O强源区。然而,我们的初步研究则显示,南大洋可能是大气N2O的汇区。本研究利用中国南极科学考察结果对南大洋的源汇特性进行分析。
1 方法
1.1 研究海域和采样方法
本研究利用中国第25、26次南极科学考察的机会,对南大洋进行走航采样工作。研究区域如图1所示,澳大利亚新西兰以南洋区及环南极60°S纬度线等南大洋区域。研究纬度范围覆盖所有南大洋典型锋面结构。
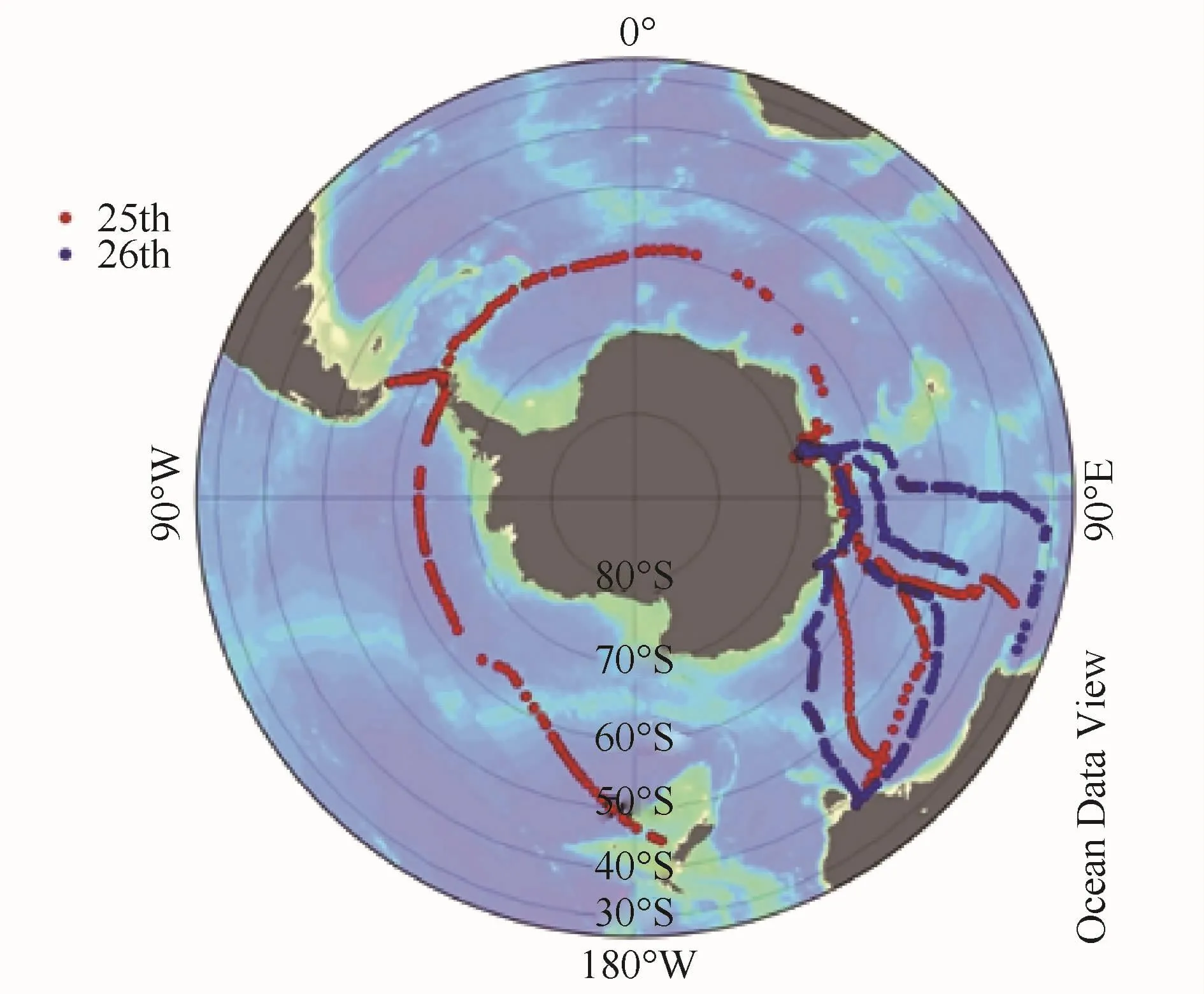
图1 中国第25、26次南极科学考察走航观测现场数据Fig.1.Cruise tracks of the 25th(red)and 26th(blue)Chinese Antarctic Research Expedition(CHINARE)
样品采集利用雪龙船配备的表层海水采集泵进行。海水采集泵进水口位于水位线下方约4.5 m处,样品采集口位于实验室内,水样全程保持流动。所采集水样N2O浓度分析结果与现场CTD水样采集获得水样分析结果无显著差异。样品使用250 mL溶氧瓶采集。所采样品添加200μL HgCl2饱和溶液后避光4℃条件保存,具体采样及保存参考Butler等[21]的方法。
1.2 分析方法
实验室使用Shimadzu GC-2010气相色谱仪进行样品分析。气相色谱配备CTC自动顶空进样器。水样分装到20 mL顶空瓶密封好后,通过自制的双针装置将一定量的水样用高纯氮气置换出来。制备好的顶空样由机械臂移至水汽平衡的振荡器平衡,达到平衡之后自动进样。色谱仪安装有专门用于分离N2O的十通阀反吹系统,柱分离系统由两根3 m长的HysepQ和一根1.9 m长的P-N柱组成。本系统可通过柱切换系统除去气体中的杂质峰,分析结果只出现N2O单峰。样品分析精度及准确度平均值均在2%左右。10 h仪器变异系数为0.6%。具体的样品分析、条件优化和参数设置见Zhan等[22]方法。
1.3 其他数据
N2O海气通量的计算利用了覆盖研究海区的遥感风速数据。该数据是由欧洲极轨气象卫星METOP-A/B卫星搭载的ASCAT散射计反演而得(ftp://ftp.remss.com/ascat/),该数据空间分辨率为0.25经度×0.125纬度。根据下载的南大洋上空10 m处的12月、1月和2月的风速数据,进一步处理成季节平均数据。
2 结果和讨论
2.1 夏季南大洋表层海水N2 O饱和度异常分布特征及调控机制
中国第25、26次南极科学考察航线表层海水表层海水温度之间的关系如图2所示。由图可见,南大洋表层海水中N2O和温度水温呈很好的指数衰减相关关系,相关系数R2=0.957,P<0.000 1,呈显著相关,说明南大洋表层海水温度是N2O分布的主控因素。该现象在Zhan等[22]的工作中曾有相关报道。然而,与上述研究不同的是,本研究具有较高的数据量和空间分布范围。研究结果显示,在亚南极峰以北表层海水中N2O浓度与平衡浓度(图2中红线所示)较为一致,说明该区域N2O与大气接近平衡;而亚南极峰以南表层水体中N2O则呈现不饱和状态。该现象十分值得关注。这是因为一直以来,由于上升流的存在,南大洋被认为是全球大气N2O重要来源区域,而本研究所覆盖较广阔的南大洋调查区域的不饱和现象却意味着南大洋至少在夏季可能是大气N2O的汇区。因此,对该区域海洋N2O源汇特征进行谨慎的分析和评估工作具有十分重要的意义。为了更加准确地分析南大洋可能存在的N2O源汇特性,计算航线上各航段表层海水的饱和度异常
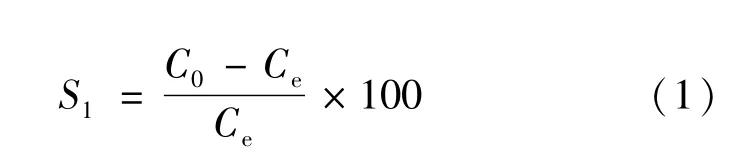
其中S1为饱和度异常值,C0和Ce分别为海水中N2O现场实测浓度和现场条件下的平衡浓度。当S1=0,表层海水中N2O与大气平衡;当S1>0时表层海水中N2O过饱和,表层海水表现为源特性,反之,当S1<0时表层海水中N2O不饱和,表层海水表现为汇特性。图3为南大洋测区表层海水饱和度异常值在不同纬度上的分布特征。由图3可见,表层海水N2O饱和度异常值北高南低,均值约在-3%,考虑分析方法存在的不确定度,可认为部分区域接近与大气的平衡,部分区域,主要是高纬度区域存在明显不饱和现象。但由于数据的精度有限,无法很清晰地区分表层海水的源汇特征差异。因此只能根据南大洋的水文特征做响应的区分。
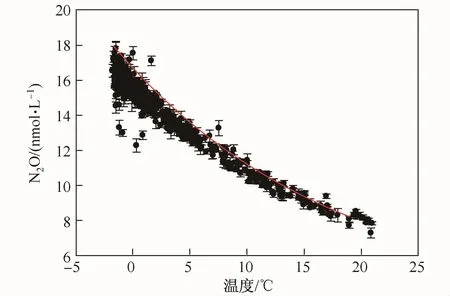
图2 中国第25、26次南极考察航线表层海水N2O浓度分布及海气平衡浓度曲线Fig.2.The surface N2O distribution patterns during the 25th and 26th CHINARE(solid circule)and the equilibrium line of surface water vesus surface water temperature(SST)(red line)
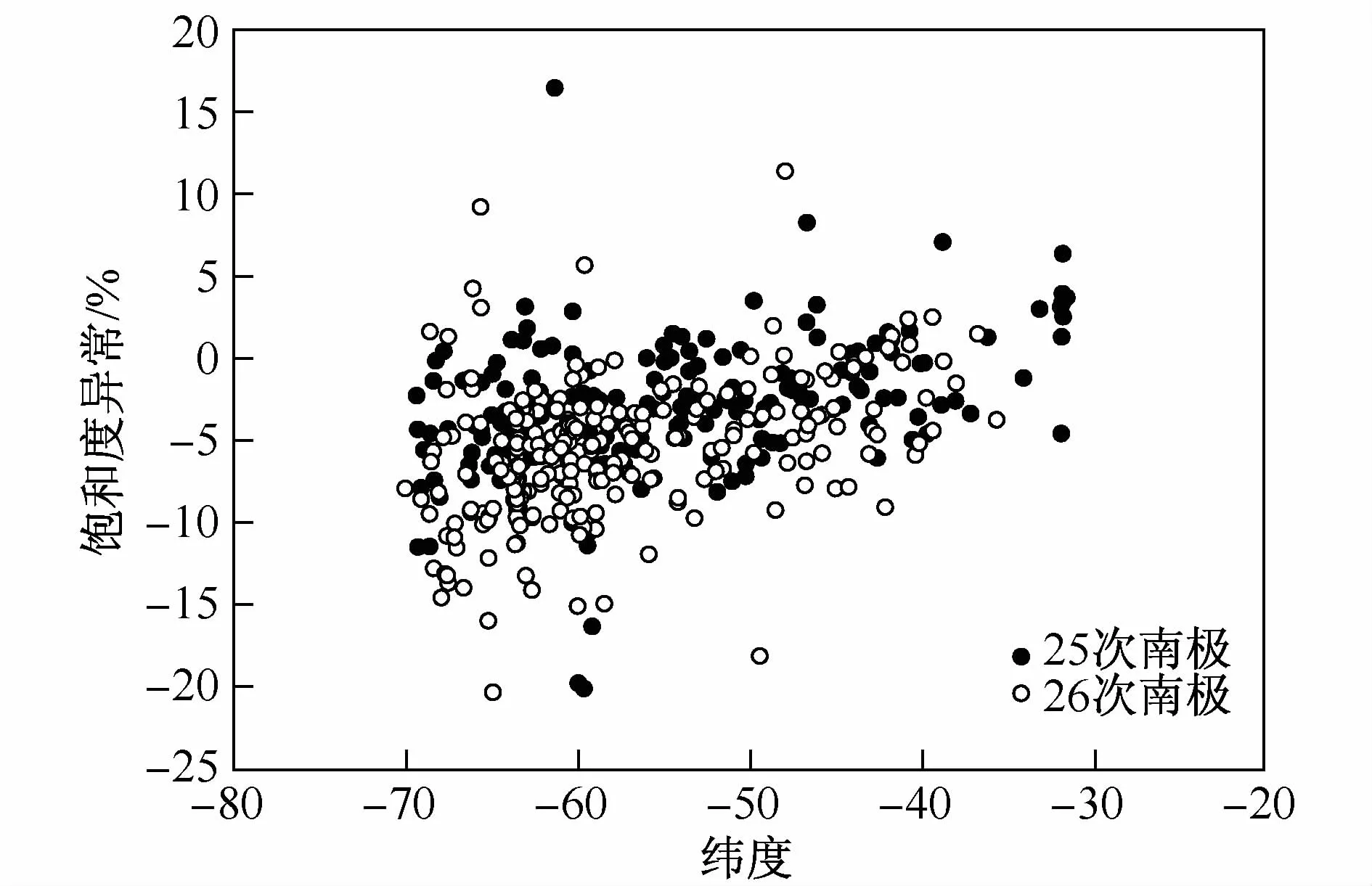
图3 南大洋表层海水饱和度异常值随纬度变化分布图Fig.3.Distribution patterns of surface N2O saturation anomally against latitude in the southern ocean during the 25th and 26th CHINARE
根据Zhan[20]的结果,N2O在南大洋的分布存在以亚南极锋(表层水温在4—8℃的快速变化区间,盐度变化为33.6—34.2)为分界线的分布特征,即亚南极锋以北显示出N2O过饱和特征,而在亚南极锋以北显示出不饱和特征,且南向增强。这种分布与南大洋水文结构特征存在相对应关系。亚南极锋以北的表层海水主要成分是亚南极模态水,该水团垂直混合均匀;而亚南极锋以南水体则为亚南极表层水,相对亚南极模态水而言,该水团呈现出低温低盐的特征,这主要是由于该水团来源于南半球高纬度表层水体的艾克曼北向运移。这一现象在25、26次南极考察中也观察到,但亚南极锋以北的饱和度异常相对Zhan等[20]的研究结果较不显著。亚南极锋以南可以观察到N2O饱和度异常值进一步降低的现象。这种变化很可能与现场水文特征有密切的关系。亚南极锋以南存在另一个锋面“极锋”,其特征为200 m以浅2℃的水温。极锋以南的表层水为南极表层水,具有低温低盐的特性。这种较低的温盐特征的形成应该是夏季融冰水对表层海水稀释作用的结果。该区域表层海水中,N2O显著的不饱和特点可能源于海冰融化对表层海水的稀释。海冰在形成过程中大部分N2O气体被“挤出”,因此所形成的海冰中N2O含量通常低于海水。海气交换过程的相对滞后自然导致了表层海水中较低N2O。上述过程可用于解释极锋以南表层海水中N2O低值形成。根据上述分析,可以得出初步结论,亚热带锋即绕极深层流的北界以北,风速较低,侧表层N2O交换速率较慢,可在表层积累并形成相对高值。亚热带锋以南和极锋之间,西风盛行,海气交换完全,表层海水N2O与大气值接近平衡;极锋以南,表层海水受融冰影响N2O呈现明显不饱和特征。
目前有限的研究均显示南大洋总体上为大气N2O来源,因此,极锋区较显著的N2O不饱和现象十分值得注意。根据上述推测,可进行如下假设,分析不饱和过程形成的机理。对至今尚未见有关南极海冰中N2O含量的报告,仅有Randall等[23]对北冰洋的海冰进行了调查,结果显示北极海冰中N2O浓度约为6 nmol·L-1。因此,以极锋为界,假设南大洋极锋以南N2O的分布特征是由N2O含量海冰融化对表层海水的稀释所致。可以根据以下混合模型,对稀释过程进行模拟。若不考虑海气交换因素,亚南极锋以南的表层海水可视为,盐度为4(PSU)的海冰端元和盐度约为34.1(PSU)(该盐度约为南极表层水最北端的盐度最高值)的表层海水端元混合而得,可得出如下混合模型:
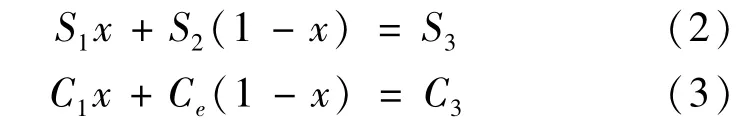
其中,S1和S2分别为海冰和海水端元的盐度,设为4和34.3,S3为混合后形成水团的盐度;x为混合水团中海冰的百分比含量;C1为海冰中N2O含量,Ce为该水团混合前该区域表层海水温度和盐度条件下表层海水中N2O的溶解度;根据各采样点的表层温盐特征和平衡浓度进行计算,获取相应采样点数据可计算出相应的C3值。所得到的C3是不考虑海气交换和表层次表层海水物质交换条件下,经过混合后相应区域浓度值。
将所获得的航迹上的CS值和现场观测C0值进行相关分析,相关结果如图4所示,两组数据呈显著相关关系(R2=0.4538,P<0.0001),说明极锋区模拟值与现场观测值显著相关。将公式1中的C0用CS代替,获得公式4。
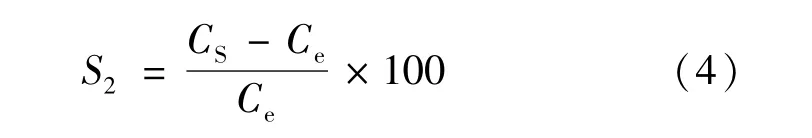
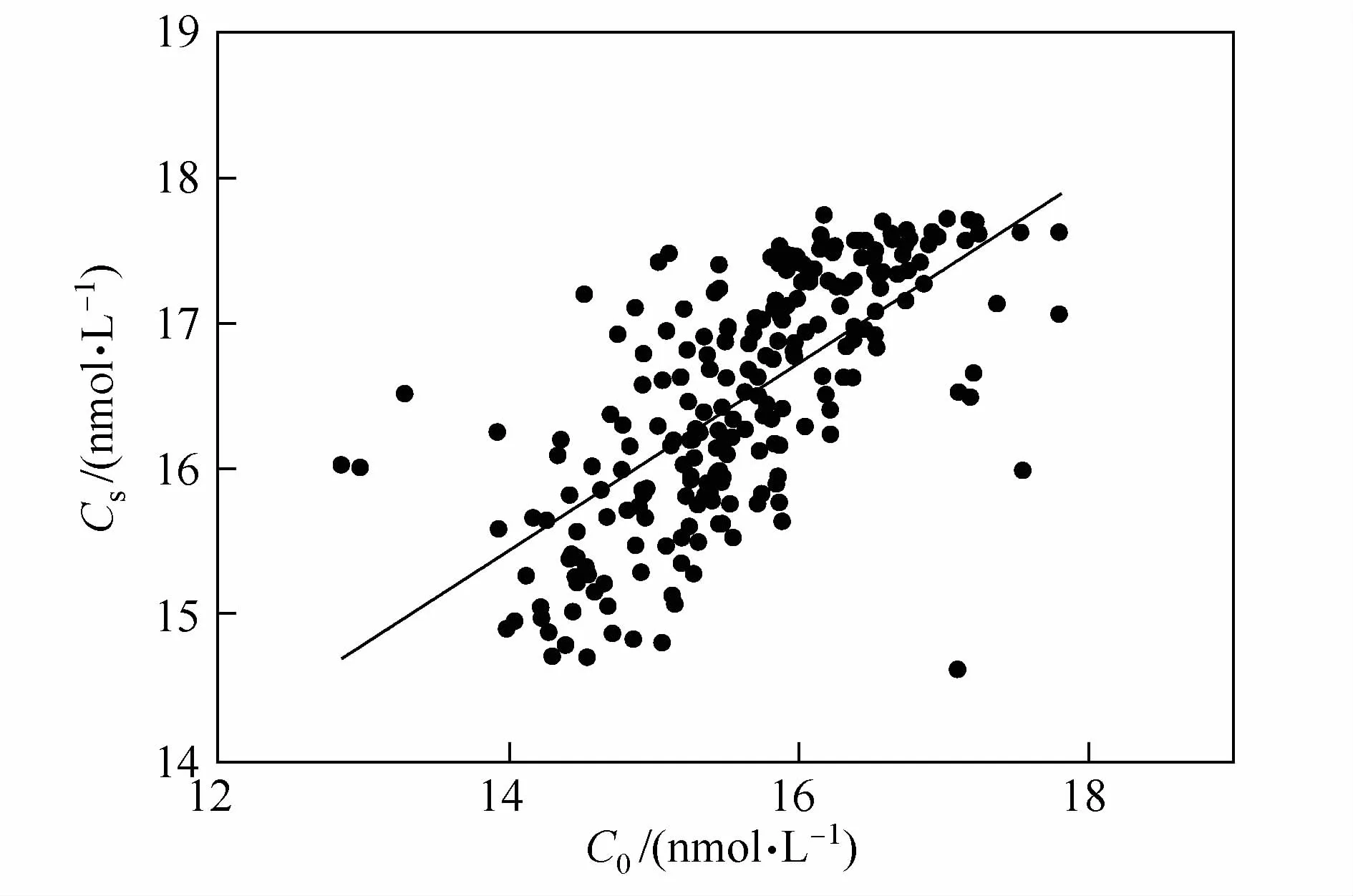
图4 南大洋亚南极锋表层海水中N2 O浓度估算值C S和观测值CO之间相关关系Fig.4.The relationship between simulated and observed N2 O concentraitons south of Polar Front(PF)in the Southern Ocean during the 25th and 26th CHINARE
该公式的意义是不考虑海气交换过程和下层水体向上输送过程的影响,仅考虑表层海水和融冰水的混合过程,混合后表层海水中N2O饱和度异常值为S2。由图5可见,极锋以南表层海水N2O饱和度异常存在离散状态,然而饱和度异常模拟值几乎都>-5%。根据计算结果显示S2和S1之间存在一定的差值。S2—S1随纬度的分布特征如图6所示。由图可得出以下信息:首先,模拟结果和现场观测结果之间差异显示,饱和度异常模拟值较现场观测值平均高约5%,单纯的混合过程无法完全解释极锋区以南表层海水的不饱和现象,表层海水不饱和的结果是大气向表层海水的传输,因此维持表层海水不饱状态应有其他的原因。其次,混合过程并非引起N2O浓度偏离平衡浓度的主要原因。可以推测,表层海水融冰过程表层海水温度变化可能是极锋以南表层海水饱和度异常偏离平衡的重要原因。
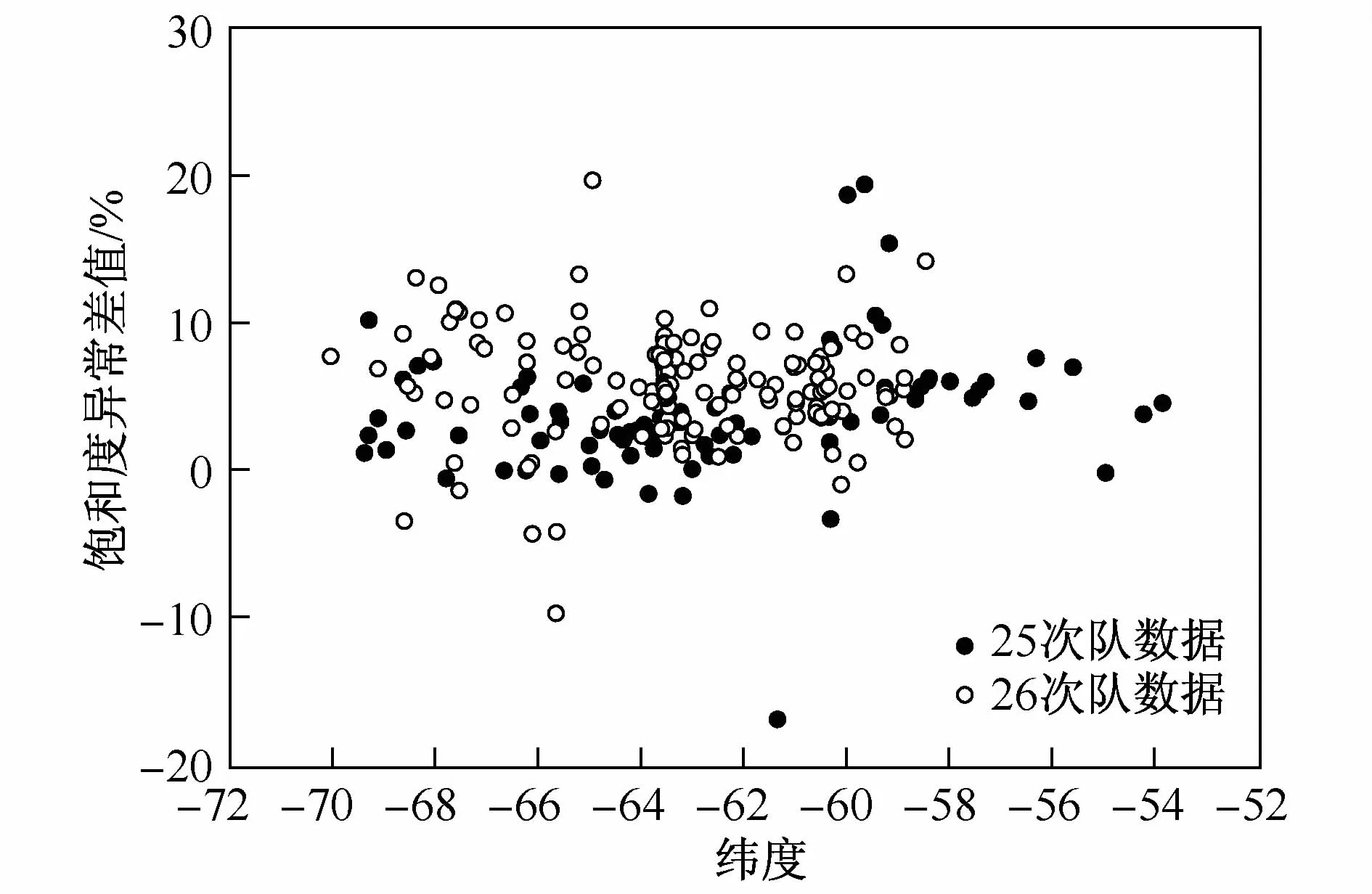
图5 极锋以南表层海水饱和度异常模拟值的纬度分布Fig.5.Distribution of simulated surface N2 O saturation anomally vesus latitude south of PF in the Southern Ocean during the 25th and 26th CHINARE
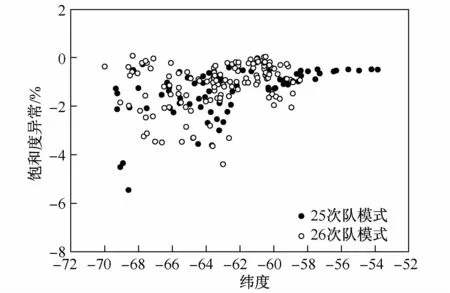
图6 极锋以南表层海水中N2O混合模型计算与观测饱和度异常值差值的纬度分布特征Fig.6.Difference between simulated N2 O surface concentrations by simple mixing model and observation vesus latitude south of PF in the Southern Ocean during the 25th and 26th CHINARE
根据现场极锋以南表层水体的分布特征,可以发现,该区域表层海水温度约在-1.9—2.0℃之间波动,并且该波动没有明显的纬度规律,导致这种变化的可能原因是海冰融化过程,海冰在融化的过程中吸收大量的热,导致表层海水温度下降。温度的波动直接改变表层海水N2O饱和度,因此如果部分区域表层海水的温度有2℃的波动,足以导致图6饱和度异常值的波动。此外,值得注意的是在航线上除了多数水体存在表层不饱和现象外,还存在少数站位存在过饱和的现象,例如61°S和66°S附近,通常与这些高值相对应的温盐数值均较周围水体高,可能与上升流的存在有密切的关系。然而,从空间分布特点上来看,夏季这种潜在的上升流在观测区域并不显著。
然而,通过上述讨论,可以发现,导致表层海水N2O变化的因素较多,并且仍然较为不确定,即使是N2O在表层海水的浓度变化仍存在较复杂的空间变化。因此,要准确评估表层海水源汇通量需要在数据空间分辨率上获得实质性的提升。此外,也需要运用遥感等相关数据评估表层海水温度变化等信息,以准确全面的分析南大洋相关区域的源汇特性和形成机制。
2.2 极锋以南洋区夏季表层海水N2 O海气通量估算
海气通量的估算是一项不确定性较高的工作,然而对于了解海洋对温室气体收支的调控作用,海气通量估算却是一项极具重要性的工作。此外,在极锋以南洋区存在的复杂的混合过程也使该项工作具有很高的挑战性。海气通量计算主要是根据海气界面存在的目标物浓度差异。可以用公式:

其中,F是海气通量,k为扩散系数,ΔC为界面浓度差。ΔC相对容易获得,而k值则通过相关的计算获得。目前已有大量的k值计算方法,Wannikhof等[24]也有相关的总结。通常的做法是通过平均风速值求得平均k值,其与区域平均浓度差即为该区域的平均通量,根据这一计算方法,运用Wannikhof等[25]可以获得极锋以南洋面海气通量约为-2.47 ±0.63 μmol·m-2·d-1,其中 0.63 μmol·m-2·d-1的误差由样品分析过程带入。

图7 极锋以南风速遥感数据Fig.7.Distribution patterns of wind speed south of PF in the southern Ocean
根据下载获得的季节风速平均数据,极锋以南表层风速数据相对平稳,随纬度增高有缓慢的增加,风速整体变化范围约在0—18.0 m·s-1(图7),然而,通过对上述风速进行正态分布分析,可以发现,整个区域95%以上的风速落在6.2—9.2 m·s-1之间。根据Wannikhof等[25]的海气通量计算公式,可以获得海气通量范围落在-3.51— -1.62μmol·m-2·d-1之间,该数据值与平均风速的计算结果显示,有分析方法引进的误差仍落在95%风速变化范围计算获得的通量范围内。由此说明,海气通量计算需要更为谨慎的处理方式。
根据上述计算的结果,可以对极锋以南洋区海气通量进行评估,假设南极极峰以南的洋区N2O和风速分布特征均通和上述描述一致,通过计算可以获得及锋以南洋区海气通量约在-3.5×10-4—-7.7×10-4Tg N·a-1。因此该区域显示为夏季大气N2O的一个弱汇。
3 结论
南大洋表层海水N2O分布特征显示亚热带锋附近表层海水显示过饱和特征,亚南极锋附近与大气接近平衡;极锋以南,表层海水呈现出一定的不饱和特征。导致该不饱和现象可能是由于海冰快速融化过程所致,快速融化过程中表层海水的温度变化对N2O不饱和的贡献较高,融冰水对N2O不饱和度的贡献则相对有限。通过已有的经验公式评估海气通量存在一定的误差,尤其在该类水域,复杂多变的表层海水特性进一步增加了评估的困难。运用正态分布统计,选取占观测风速95%以上的风速范围进行海气通量评估。所得结果显示,由于风速变化不确定度带来的风速变化高于实验分析误差带来的不确定度。极锋以南洋区是大气N2O的汇区,年贡献量为-3.5—-7.7×10-4百万吨氮。要更准确地了解极锋区以南洋区的不饱和现象以至整个南大洋的分布特征,需要在观测手段上有所提高,以获得更高分辨率的数据。同时,通过结合遥感表层海水温度、盐度和海冰等信息仪器获得对相关区域更为精确的通量评估结果。
1 Crutzen P J.The influence of nitrogen oxides on the atmospheric ozone content.Quarterly Journal of the Royal Meteorological Society,1970,96(408):320—325.
2 Ravishankara A R,Daniel JS,Portmann RW.Nitrous oxide(N2O):The dominant ozone-depleting substance emitted in the 21st century.Science,2009,326(5949):123—125.
3 Junge C,Hahn J.N2O measurements in the North Atlantic.Journal of Geophysical Research,1971,76(33):8143—8146.
4 Hahn J.The North Atlantic Ocean as a source of atmospheric N2O.Tellus,1974,26(1-2):160—168.
5 Hahn J,Thrush B A,Nash T.The cycle of atmospheric nitrous oxide.Royal Society of London Philosophical Transactions Series A,1979,290(1376):495—504.
6 Hahn J.Nitrous oxide in the oceans//Delwich CC.Denitrification,Nitrification,and Atmospheric Nitrous Oxide.New York:John Wiley&Sons Inc,1981:191—277.
7 Codispoti L A,Christensen JP.Nitrification,denitrification and nitrous oxide cycling in the eastern tropical South Pacific Ocean.Marine Chemistry,1985,16(4):277—300.
8 Cohen Y,Gordon L I.Nitrous oxide production in the ocean.Journal of Geophysical Research,1979,84(C1):347—353.
9 Elkins JW,Wofsy SC,McElroy M B,et al.Aquatic sources and sinks for nitrous oxide.Nature,1978,275(5681):602—606.
10 Oudot C,Andrie C,Montel Y.Nitrous oxide production in the tropical Atlantic Ocean.Deep-Sea Research Part A.Oceanographic Research Papers,1990,37(2):183—202.
11 Ding Y,Griggs D J,Noguer M,et al.Climate Change2001:The Scientific Basis.Cambridge:Cambridge University,2001.
12 Nevison CD,Weiss R F,EricksonⅢ D J.Global oceanic emissions of nitrous oxide.Journal of Geophysical Research,1995,100(C8):15809—15820.
13 Suntharalingam P,Sarmiento JL,Toggweiler JR.Global significance of nitrous oxide production and transport from oceanic low-oxygen zones:A modeling study.Global Biogeochemical Cycles,2000,14(4):1353—1370.
14 Lo Monaco C,Metzl N,Poisson A,et al.Anthropogenic CO2in the southern ocean:distribution and inventory at the Indian-Atlantic boundary(World Ocean Circulation Experiment line I6).Journal of Geophysical Research,2005,110(C6):doi:10.1029/2004JC002643.
15 Priscu JC,DownesM T,Priscu L R,etal.Dynamicsofammonium oxidizer activity and nitrous oxide(N2O)within and beneath Antarctic sea ice.Marine ecology progress series.Marine Ecology Progress Series,1990,62(1-2):37—46.
16 Weiss R F,Van Woy F A,Salameh PK,etal.Surfacewater and atmospheric carbon dioxide and nitrous oxide observationsby shipboard automated gas chromatography:Results from expeditions between 1977 and 1990.Washington,DC:Oak Ridge National Laboratory,TN(United States),Carbon Dioxide Information Analysis Center,1992.
17 Rees A P,Owens N P,Upstill-Goddard R C.Nitrous oxide in the Bellingshausen Sea and Drake Passage.Journal of Geophysical Research,1997,102(2):3383—3392.
18 Bouwman A F,van der Hoek KW,Olivier JG J.Uncertainties in the global source distribution of nitrous oxide.Journal of Geophysical Research,1995,100(D2):2785—2800.
19 Law CS,Ling R D.Nitrous oxide flux and response to increased iron availability in the Antarctic Circumpolar Current.Deep-Sea Research PartⅡ:Topical Studies in Oceanography,2001,48(11-12):2509—2527.
20 Zhan L Y,Chen L Q.Distributions of N2O and its air-sea fluxes in seawater along cruise tracks between 30°S—67°Sand in Prydz Bay,Antarctica.Journal of Geophysical Research,2009,114(C3):1—14.
21 Butler JH,Elkins JW.An automated technique for themeasurementof dissolved N2O in naturalwaters.Marine Chemistry,1991,34(1-2):47—61.
22 Zhan L Y,Chen L Q,Zhang JX,et al.A system for the automated static headspace analysis of dissolved N2O in seawater.International Journal of Environmental Analytical Chemistry,2012,93(8):828—842.
23 Randall K,Scarratt M,Levasseur M,et al.First measurements of nitrous oxide in Arctic sea ice.Journal of Geophysical Research:Oceans(1978—2012),2012,117(C9):doi:10.1029/2011JC007340.
24 Wanninkhof R,AsherW E,Ho D T,et al.Advances in quantifying air-sea gas exchange and environmental forcing.Annual Review ofMarine Science,2009,1(1):213—244.
25 Wanninkhof R.Relationship between wind speed and gas exchange over the ocean.Journal of Geophysical Research,1992,97(25):7373—7382.
