正极集流体对可充镁电池电化学性能的影响
2015-01-04苏硕剑努丽燕娜非路热吐尔逊王久林上海交通大学化学化工学院上海200240
苏硕剑 努丽燕娜 非路热·吐尔逊 杨 军 王久林(上海交通大学化学化工学院,上海200240)
正极集流体对可充镁电池电化学性能的影响
苏硕剑 努丽燕娜*非路热·吐尔逊 杨 军 王久林
(上海交通大学化学化工学院,上海200240)
以目前常用的Chevrel相Mo6S8作为正极材料,涂覆在不同集流体(不锈钢、镍、铜、钛)上,以镁为负极,研究了在(PhMgCl)2-AlCl3/四氢呋喃(简称THF)“二代”电解液中集流体对可充镁电池电化学性能的影响.恒流放电-充电结果显示在不锈钢集流体上电池电压极化最小,并且具有较好的循环稳定性;镍、铜次之;钛集流体上的极化最大,循环稳定性也最差.并通过对比放电-充电循环前后电极和集流体表面的微观结构,探讨了集流体对电池性能显著影响的原因.电解液对集流体会造成腐蚀,不同集流体在电解液中的稳定性有差异;正极材料涂覆在不同集流体上,电极表面状况有差异;负载活性材料后集流体发生腐蚀的电位有所降低,使集流体更易受到电解液的腐蚀.
可充镁电池;正极;集流体;电化学性能;腐蚀
1 引言
锂离子电池以其独特的优点在各种手持设备和笔记本电脑等电子产品中得到大量的应用,但在大容量储电、电动车用电池方面,可充镁电池拥有着锂离子电池不可比拟的优势:其一,镁在地壳中的储量丰富,远远超过了锂的储量,这使得金属镁的价格远低于锂的价格(镁和锂分别为$2700/吨和$64800/吨);其二,镁的化学性质没有锂活泼,加工操作更安全.且不易产生枝晶,安全性高;其三,镁具有比锂更高的理论体积比容量(分别为3832和2062 mAh·cm-3),在可安装电池的有限空间方面更具有竞争力.1-3基于以上几个原因,可充镁电池具有成为高容量、大输出功率的动力电池的潜力.
©Editorial office of Acta Physico-Chimica Sinica
一直以来,人们将可充镁电池的研究重点放在正极材料和电解液两个方面.4-7目前电化学性能稳定的正极材料是Chevrel相的Mo6S8,理论比容量为122 mAh·g-1;8,9常用的电解液是Aurbach等开发的铝基电解液体系:由Lewis碱Mg(Bu)2和Lewis酸AlEtCl2反应得到的0.25 mol·L-1Mg(AlCl2BuEt)2/四氢呋喃(以下简称THF)“一代”电解液,其在Pt电极上的阳极氧化分解电位接近2.5 V(vs Mg/Mg2+);10-12由Lewis碱PhMgCl和Lewis酸AlCl3按摩尔比2:1在THF中反应制得0.40 mol·L-1(PhMgCl)2-AlCl3/THF“二代”电解液,其在Pt电极上的阳极氧化分解电位可达到3 V(vs Mg/Mg2+).13-152011年,Kim等3首次报道了非亲核电解液[Mg2Cl3-6THF][HDMSAlCl3](其中HDMS为(Me3Si)2N),其氧化分解电位可达3.2 V (vs Mg/Mg2+),并初步研究了与硫正极材料的匹配性能.近年来一种基于有机硼镁盐的电解液被相继提出,其具有3.5 V(vs Mg/Mg2+)高的电化学稳定性,高的离子电导率以及优良的可逆沉积-溶出镁的特性.16,17我们课题组18报道了一系列酚基电解液,这些电解液具有对空气不敏感、过电位低的优点,在Pt电极上的阳极氧化分解电位为2.6 V(vs Mg/Mg2+).这些电解液中,与正极材料Mo6S8匹配性良好,并且研究广泛透彻的为“一代”、“二代”电解液.研究中发现,由于这两种电解液的有机镁盐的氯化物组分中均含有氯离子,会对常用作集流体的金属基质产生腐蚀.19Lv等20使用线性电位扫描法研究了铜、镍、不锈钢、铝、钛、铂作为集流体在“一代”电解液中的电化学稳定性,发现它们具有不同的氧化分解电位.Yagi等21使用循环伏安法研究了非贵金属集流体(铜、镍、钛、铝、不锈钢)在“二代”电解液中的电化学稳定性,并使用扫描电子显微镜(SEM)观测了集流体的表面形貌变化.我们课题组22系统研究了“一代”、“二代”电解液在铂、镍、不锈钢、铜、铝五种金属基质上的电化学性能,发现电解液的阳极氧化分解电位和镁沉积-溶出性能在不同金属上有明显差异,确定了这些金属在“一代”、“二代”电解液中发生腐蚀的电位.
目前可充镁电池的负极通常使用金属镁(合金)片,可以不用集流体.由于电解液在不同集流体上的电化学性能不同,那么正极材料所用的集流体则可能对正极材料以至整个电池的性能产生显著影响.随着宽电化学窗口电解液和高工作电压正极材料的开发和研究,确定实用、廉价、耐腐蚀的正极材料集流体是可充镁电池研究中不容忽视的问题.本文选择不锈钢箔、镍箔、钛箔、铜箔等常用集流体作为研究对象,以Chevrel相硫化物Mo6S8作为正极材料,镁为负极,“二代”电解液为电解液,组装电池,测试其电化学性能,系统研究正极集流体对可充镁电池性能的影响.
2 实验部分
2.1 试剂与仪器
MoS2(99%)、Mo粉(99%)、KCl(99%)、镁条(99.5%)以及苯基氯化镁(2.0 mol·L-1的四氢呋喃溶液)购于Sigma-Aldrich,CuS·H2O(99.0%)购于阿拉丁,不锈钢箔、镍箔购于盛大金属有限公司,钛箔购于上海镇铭有色金属材料有限公司,铜箔购于联鑫科技铜箔有限公司,无水三氯化铝(AlCl3(99.99%))购于TCI,无水四氢呋喃(THF)由实验室自组装设备重蒸纯化制得.对水敏感的试剂放置于氩气气氛手套箱(MBRAUN,德国UNILAB)中备用.不锈钢(SS)、镍(Ni)、铜(Cu)、钛(Ti)盘(d=2 mm)电极购买于上海仙仁仪器仪表有限公司.
实验扣式电池(CR2016)的组装在氩气气氛手套箱中进行.CHI660C电化学工作站(上海辰华仪器有限公司)上进行循环伏安扫描测试,LANDCT2001A系统(武汉蓝电电子有限公司)测试扣式电池的电化学性能.X射线衍射仪(D/max-2200/PC,Japan Rigaku Corporation)测定合成的Mo6S8组分,场发射扫描式电子显微镜(JSM-7401F,JEOL Ltd.,Japan)拍摄集流体的形貌.
2.2 实验方法
2.2.1 电解液的配制
在氩气气氛手套箱内,将苯基氯化镁与无水三氯化铝按照2:1的摩尔比混合搅拌,加入适量无水THF配制成黄色透明的0.40 mol·L-1(PhMgCl)2-AlCl3/THF电解液.
2.2.2 Mo6S8正极材料的制备
首先通过熔盐法合成Chevrel相Cu2Mo6S8,反应如(1)式所示:23
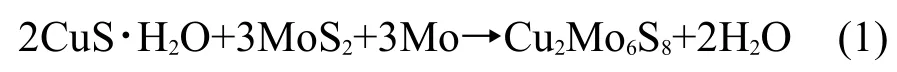
按照化学计量比称取CuS·H2O、MoS2、Mo粉以及熔盐KCl(反应物与熔盐的摩尔比为1:2),研磨使其均匀混合,在氩气气氛保护下升温至850°C温度下烧结60 h,升温速率为2°C·min-1,然后自然冷却至室温.所得粉末用热的去离子水超声清洗3-4次,离心后110°C下真空干燥2 h,得到Cu2Mo6S8.
将1.2 g的Cu2Mo6S8材料浸渍在20 mL 6 mol· L-1的盐酸水溶液中,在室温下搅拌2-4天,过程中通7 h O2(100 cm3·min-1)以加速反应进行,其反应方程式如式(2)所示;24之后将该溶液离心,用100 mL去离子水清洗3-4次,离心后120°C下真空干燥2 h,得到Mo6S8正极材料.

2.2.3 电化学性能的测试
电解液中镁的电化学沉积-溶出性能和氧化分解电位通过循环伏安曲线(CV)来测定,测试过程均在室温、氩气手套箱中进行.实验采用三电极体系,工作电极使用前用三氧化铝粉末进行抛光处理,然后用去离子水及无水乙醇清洗,烘干后使用.参比电极、对电极均采用表面用800 Cw砂纸打磨处理干净的镁条.测试用的三电极管及塞子均经过烘干处理.测量时,从开路电位开始向负方向扫描,扫描速率为50 mV·s-1.
按质量比8:1:1称取正极活性材料、Super-P导电碳和聚偏二氟乙烯(PVDF)粘结剂(PVDF预先溶于N-甲基吡咯烷酮(NMP)中配成浓度0.02 g·mL-1的溶液)置于小烧杯中,磁力搅拌4 h得到混合均匀的粘稠浆料,将浆料均匀涂覆于不锈钢箔、镍箔、钛箔或铜箔集流体上,在80°C下干燥除去溶剂后,冲成均一的极片;在120°C下真空干燥5 h后,迅速转移至氩气手套箱中,以打磨光亮的镁条为负极,Entek PE膜为隔膜,与“二代”电解液一起组装成CR2016扣式可充镁电池.组装好的电池在室温下静置4 h,使电解液完全浸润后进行放电-充电性能测试.通过CR2016扣式电池的恒电流放电-充电测试不同集流体上Mo6S8正极材料在“二代“电解液中的性能.测试条件采用0.05C(1C=122 mA·g-1)倍率,放电截止电压为0.50 V(vs Mg),充电截止电位根据采用的集流体在变化,具体在下文中说明.
2.2.4 XRD及SEM测试
采用X射线衍射(XRD)表征制备好的Mo6S8的组分,测试中使用Cu靶的Kα射线(0.154056 nm)进行实验,扫描速率为5(°)·min-1.采用扫描电子显微镜分析放电-充电后集流体上正极材料表面和集流体发生变化的形貌,样品制备采用与正常扣式电池相同的程序,电池以0.05C倍率的电流进行放电-充电,达到一定条件后,将电池在手套箱内拆开,将集流体上的正极材料表面电解液用THF清洗干净;另将集流体上的正极材料用乙醇浸泡,超声除去,并用THF清洗干净;后用滤纸将THF吸干,进行扫描电子显微镜测试,样品不需要喷金预处理.
3 结果与讨论
3.1 Mo6S8的XRD表征
测试正极集流体的性能时,正极材料的选择至关重要,具有高充放电电压平台、高容量、循环性能稳定的正极材料是首选.研究中发现,现有报道的可充镁电池电解液大多会对作为常用集流体的金属(如铜、铝、不锈钢、镍等)产生腐蚀,大大降低其电化学窗口.因而,虽然Mo6S8正极材料放电-充电电压平台较低(在1.20、1.00 V左右处有两个放电平台,在1.30、1.60 V左右处有两个充电平台),但其在现有各种电解液中电化学性能稳定,是目前作为研究正极集流体的最佳材料.
Mo6S8是一种热力学不稳定的材料,仅可由较稳定、含有金属的Chevrel相如Cu2Mo6S8通过化学或者电化学浸渍除去铜离子的方法制备.与电化学浸渍方法(为了避免铜离子对镁表面的负面影响,需要将Cu2Mo6S8与镁放在不同的电解池中)相比,化学浸渍在技术上更加可行.其中通过在HCl/H2O中化学浸渍的方法可以得到电化学性能优异的Mo6S8材料.24
图1(a)为合成产物的XRD图谱,除了目标产物Mo6S8(JCPDS-ICDD,27-0319)的衍射峰之外,出现了反应物MoS2(JCPDS-ICDD,65-0160)的衍射峰,说明所合成的样品中含有未反应的MoS2.通过熔盐方法合成Mo6S8时,不可避免会在合成产物中出现MoS2杂质相,也会有MoO2(JCPDS-ICDD,65-1273)存在.24实验证明,通过延长Cu2Mo6S8在HCl/H2O中化学萃取的时间,可以降低目标产物Mo6S8中MoS2杂质相的含量.为了确定合成产物中杂质的含量,我们对此XRD图谱进行了Rietveld全谱拟合(软件为highscoreplus),结果如图1(b)所示.采用无标定量分析可得Mo6S8、MoS2和MoO2的含量分别为77.19%、18.15%和4.66%(质量分数).由于我们在后续集流体性能的测试时,采用同一批合成的Mo6S8,得到的数据具有可比性.
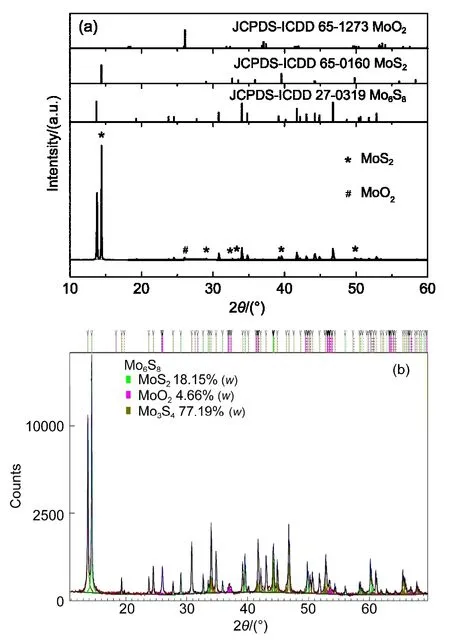
图1 熔盐方法合成的Mo6S8的XRD图谱(a)以及相应的Rietveld拟合结果(b)Fig.1 XRD pattern(a)and Rietveld refinement(b)of Mo6S8synthesized by a molten salt method
3.2 Mo6S8/Mg电池在不同集流体上的电化学性能比较
为了确定Mo6S8中含量较多的杂质相MoS2对电池性能的影响,我们把原料中的MoS2制成极片(集流体为不锈钢),组装电池测试其放电-充电性能,结果如图2所示,可以看出MoS2的活性极低,放电、充电容量不足1 mAh·g-1,其对Mo6S8性能的影响可忽略不计.
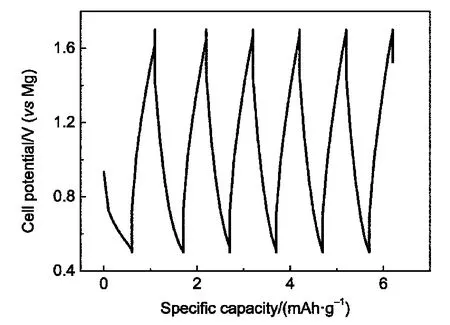
图2 MoS2/Mg扣式电池的循环放电-充电曲线Fig.2 Discharge-charge cycling curves of a MoS2/Mg coin cell
图3为在不同集流体上Mo6S8/Mg电池循环过程中的放电容量.在不锈钢、镍、铜、钛这四种集流体上Mo6S8均具有较高的首次放电容量,其中镍、钛、铜、不锈钢上为108、99、89和73 mAh·g-1.均与Mo6S8的理论容量122 mAh·g-1有差距,主要是因为合成的产物中含有一定量的杂质.相比于首次放电容量,循环过程中第二次放电容量都有大的降低,其中镍只有第一次的61%,铜、钛和不锈钢分别降低到第一次的68%、70%和93%.Mo6S8在这四种集流体上均具有首次容量损失,这种容量损失在镍上表现得最为明显.首次容量损失(约20%-25%)主要来自于镁在Mo6S8中首次脱出过程不完全,也与浸渍过程中材料表面形成导电性较低的膜阻碍了Mg的脱出有关.24循环20次后,不锈钢上的容量保持率为首次的67%,镍、铜和钛上的容量保持分别为首次的52%、50%和20%,Mo6S8正极材料在不锈钢上表现出最好的循环稳定性,而在钛上循环稳定性最差.由于所用Mo6S8为同一批制备的材料,出现的容量损失应与所用的集流体有关.电极制备过程中, Mo6S8材料在四种集流体上涂覆均匀牢固,肉眼看来没有差别.活性物质浆料与金属集流体的结合力决定了涂覆层的表面状态,其在这四种金属上应该有所差异.图4为不锈钢、镍、铜、钛四种集流体上Mo6S8电极未参加反应和循环过后的SEM.与不锈钢、铜上较为致密均匀的涂覆层相比,镍、钛上的涂覆层较为松散,会导致电流分布不均匀;也会使集流体与活性物质接触不充分,活性物质利用率降低.循环过后,不同集流体上活性物质颗粒都均匀细化.与不锈钢相比,镍、特别是钛上活性物质表面均匀性欠佳,铜上表面层则出现了明显的孔洞,可能是其循环性能较差的原因之一.
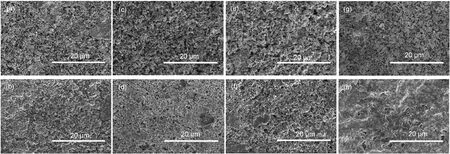
图4 不同电极片放电-充电循环测试前后的SEM图Fig.4 Scanning electron microscope(SEM)images of different electrodes before and after discharge-charge cycling measurements
图5为在不同集流体上Mo6S8/Mg电池循环第三次和第十次的放电-充电曲线.在这四种集流体上都有两个放电-充电平台,一个长而平,分别在1.00和1.30 V(vs Mg)左右,另一个较不明显,在1.20和1.60 V(vs Mg)左右,这与文献1中报道的相符.在Mo6S8晶体结构中,Mo原子的八面体簇位于S原子的立方体中,相邻的两个Mo6S8单元之间有可供镁原子进入的通道,原则上有12个位置可以嵌入镁,但由于空间和静电场的影响,仅有一些位置可被镁同时占据.Mg的嵌入过程分两个步骤进行,第一步时Mg2+占据内部位置,对应着高电压的放电平台;第二步时Mg2+占据外部位置,对应低电压的放电平台.当Mg脱出时,由于两个Mg2+之间存在斥力,第二步的Mg2+容易脱出,对于低电压的充电平台;第一步的Mg2+较难脱出,对应于高电压的充电平台.从图中可以看到Mo6S8在不锈钢上的电压极化(放电-充电平台电位差)最小,为0.10 V,而且平台稳定,后期慢慢增大至0.20 V;在镍上电压极化为0.20 V左右,随循环变化不大;在铜上亦是如此;而在钛上电压极化从一开始的0.20 V随循环增大到0.30 V左右,相比于其它三种集流体,其平台稳定性最差.可以看到,集流体对可充镁电池的电化学性能有显著的影响.

图5 不同集流体Mo6S8/Mg扣式电池第三次和第十次的放电-充电曲线Fig.5 The third and tenth discharge-charge curves for Mo6S8/Mg coin cells with different current collectors
3.3 不同集流体的腐蚀电位
目前测试可充镁电池电解液的电化学窗口(特别是氧化分解电位)一般使用惰性电极如铂电极,可以避免电极和电解液之间的反应,确保在电极上发生的电化学反应仅仅与电解液的分解有关.但在实际使用过程中不会用铂作为集流体,因而在非惰性电极即常用作集流体的金属上电解液的氧化分解电位更加实用,但此时发生的电化学反应不仅与电解液的氧化分解有关,也会涉及到金属电极与电解液的反应(如金属被腐蚀)以及电极自身的氧化,这里统称为稳定电位.图6为使用三电极体系测得的不锈钢、镍、铜、钛电极上“二代”电解液的循环伏安曲线,对电极和参比电极为镁条.在几种电极上都可以进行镁的电化学沉积和溶出.在从开路电位向负向扫描的过程中,从-0.20 V(vs Mg/Mg2+)以下开始出现沉积电流,沉积过程中呈现一个典型的由过电位驱动产生晶核形成及晶核生长的成核环;正向扫描过程中,沉积镁的溶出从0.00 V(vs Mg/Mg2+)左右开始,在0.50 V(vs Mg/Mg2+)以上出现镁的氧化溶出峰;再向正电压方向扫描时电流开始基本保持不变,表明没有副反应发生,直到电解液发生分解,刚开始出现电流明显上升的电位即为稳定电位.从中可以得出“二代”电解液中铜、不锈钢和镍电极上的稳定电位分别为1.88、2.21和2.22 V(vs Mg/Mg2+).对于钛电极而言,在正向扫描过程中电解液的稳定电位可达到2.45 V(vs Mg/Mg2+),回扫过程中出现回扫电流大于正向扫描电流的现象(见图6插图),说明钛表面发生了腐蚀的过程,导致极化电流逐渐增大;25但第二次正向扫描过程中并没有因为出现的腐蚀现象使稳定电位有所降低,分解电流也保持不变,说明腐蚀现象发生的同时也在钛电极表面形成了表面膜(从下面的SEM测试结果中可以看出),使得第二次循环时钛电极的稳定电位保持不变.
当将这些金属作为集流体组装到扣式电池中时,由三电极得到的阳极稳定电位和两电极(如扣式)实际电池的充电截止电压应该是不同的.一般认为,后者低于前者100 mV左右.16那么实际电池中铜、不锈钢、镍和钛集流体的稳定电位应该分别为1.78、2.11、2.12和2.35 V左右.

图6 不同电极上0.40 mol·L-1(PhMgCl)2-AlCl3/THF的循环伏安曲线Fig.6 Cyclic voltammogram curves of different electrodes in 0.40 mol·L-1(PhMgCl)2-AlCl3/THF
为了研究实际电池体系中集流体的稳定性,我们把加入粘结剂、导电碳的正极材料涂覆在上述四种不同的集流体上,制成极片,组装成扣式电池后进行恒电流放电-充电测试.充电截止电压为1.70-2.10 V,首次为1.70 V,之后每循环一次增加0.10 V,直至充电截止电压为2.10 V,所得结果如图7所示.
从图中可以看出在镍上充电截止电压从1.70 V一直到2.00 V电池都可以正常进行充放电;当充电截止电压升至2.10 V后,电池电压充到2.08 V之后就降下来,之后一直维持在1.94 V左右,始终充不到充电截止电压2.10 V.在不锈钢上充电截止电压从1.70 V到2.00 V,电池都可以正常循环;当充电截止电压升至2.10 V,电池循环了3次后就再充不到2.10 V,之后一直维持在1.93 V左右.在铜上,充电截止电压为1.70和1.80 V时电池可以正常进行充放电;充电截止电压升到1.84 V后,出现了个小小的平台;充电截止电压升到2.00 V后,电池充到1.98 V左右就开始慢慢降至1.86 V左右,最低处可达到1.70 V.在钛上充电截止电压从1.70到1.90 V电池都可以正常进行充放电;当充电截止电压升至2.00 V时,电池电压充到1.91 V之后就衰减下来,之后在1.58 V左右维持了一段时间,后又降到了1.40 V.四种集流体上均出现随时间的延续充电电压持续降低且不稳定的现象,这种现象应该与集流体被腐蚀的过程有关.
为了得到证实,我们将上面的电池拆开,用乙醇浸泡超声除去活性材料Mo6S8、粘结剂和导电剂,得到表面干净的集流体;进一步用THF溶剂清洗,干燥后进行SEM测试,观察其表面形貌;为了比较,将新鲜的镍、不锈钢、铜、钛箔也做了SEM测试,结果如图8所示.可以看到四种集流体表面都发生了不同程度的腐蚀.循环放电-充电后光滑的镍、不锈钢箔表面都可以看到明显的蚀坑存在;而所用的铜、钛箔表面是粗糙的,具有颗粒状的形貌,放电-充电后则可观察到表面粗糙部分被腐蚀掉.这表明这些金属作为可充镁电池的集流体使用时,达到一定充电电压后除了发生电解液自身的分解外,也伴随有金属自身的腐蚀.这种腐蚀很大程度上与电解液有机镁盐的氯化物组分中存在的Cl-有关,由于Cl-的半径小、穿透力强,故它很容易穿透金属表面保护膜有缺陷的地方,与金属离子结合成可溶性离子,随着金属离子的不断溶解,在金属表面形成了点蚀坑.26为了减少电解液对集流体的腐蚀,目前的研究逐渐趋向于采用无卤素离子的有机镁盐27或者采用无机镁盐如Mg(BH4)228,29体系.
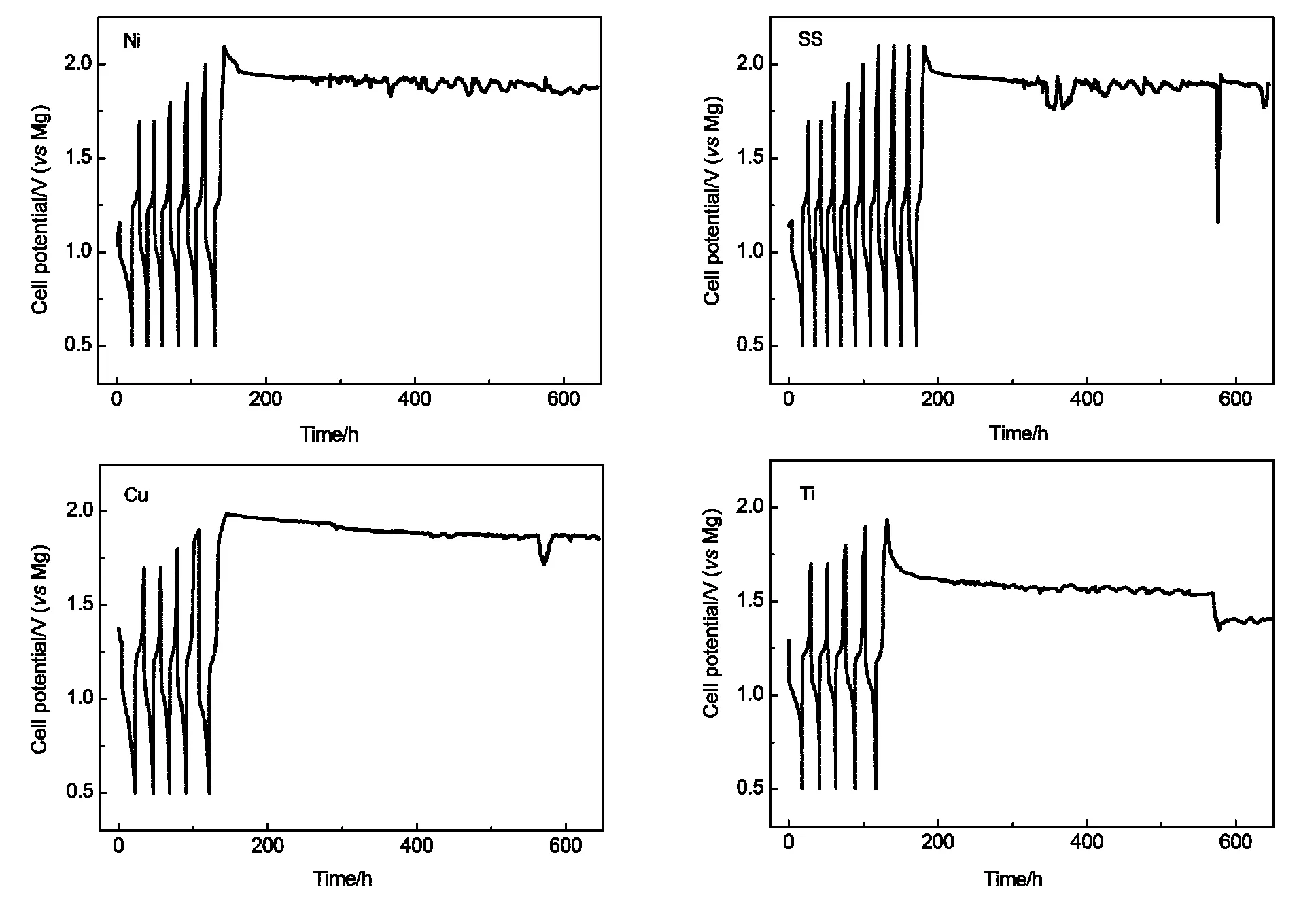
图7 不同集流体Mo6S8/Mg扣式电池的循环放电-充电曲线Fig.7 Discharge-charge cycling curves of Mo6S8/Mg coin cells with different current collectors

图8 Mo6S8/Mg扣式电池放电-充电测试前后不同集流体除去负载材料的表面SEM形貌图Fig.8 SEM images of different current collectors removing loading material before and after discharge-charge measurements for Mo6S8/Mg coin cells
以上研究表明,实际电池体系中的集流体的稳定性比三电极体系中的有所降低,其中钛从2.45 V降到了1.40 V,铜从1.88 V降到了1.70 V,不锈钢从2.21 V降到了1.93 V,镍从2.22 V降到了1.94 V.而与前面提到的实际电池中钛、铜、不锈钢和镍集流体的稳定电位应该分别为2.35、1.78、2.11和2.12 V左右有较大的差距.可以考虑到的是在实际电池体系中集流体表面涂覆了正极材料、粘结剂和导电碳,所处的环境与单一的集流体不一样.除去电阻产生的较小极化之外,可能是正极材料、粘结剂或导电碳对集流体的腐蚀起了一定的促进作用.
为了弄清楚实际电池体系中集流体腐蚀电位为什么会降低,我们把没有负载活性材料的这四种集流体作为正极装配成扣式电池进行镁沉积-溶出测试,充电截止电压为0.80-2.40 V,前三次充电截止电位为0.80 V,先使镁沉积-溶出稳定,然后升到1.70 V,之后每循环一次增加0.10 V,直至出现电压充不上去或远远高于沉积的镁溶出的电量,所得结果如图9所示.可以看到,铜集流体在1.80 V以上开始出现电压不稳定、镁溶出电量高于沉积电量的情况,表明出现了铜的腐蚀.不锈钢集流体可以充电到2.20 V,但不能持续循环,也出现电压不稳定、镁溶出电量高于沉积电量的情况,对应于不锈钢的腐蚀.镍集流体到2.00 V特别是2.10 V时出现镁溶出电量高于沉积电量的情况,为镍的腐蚀.而Ti集流体可以稳定充电到2.40 V,才出现前面三种集流体上出现的与腐蚀对应的现象.这与前面提到的实际电池中铜、不锈钢、镍、钛集流体的稳定电位分别为1.78、2.11、2.12和2.35 V左右是相符合的.铜较低的稳定性限制了其作为高电压正极材料的集流体.
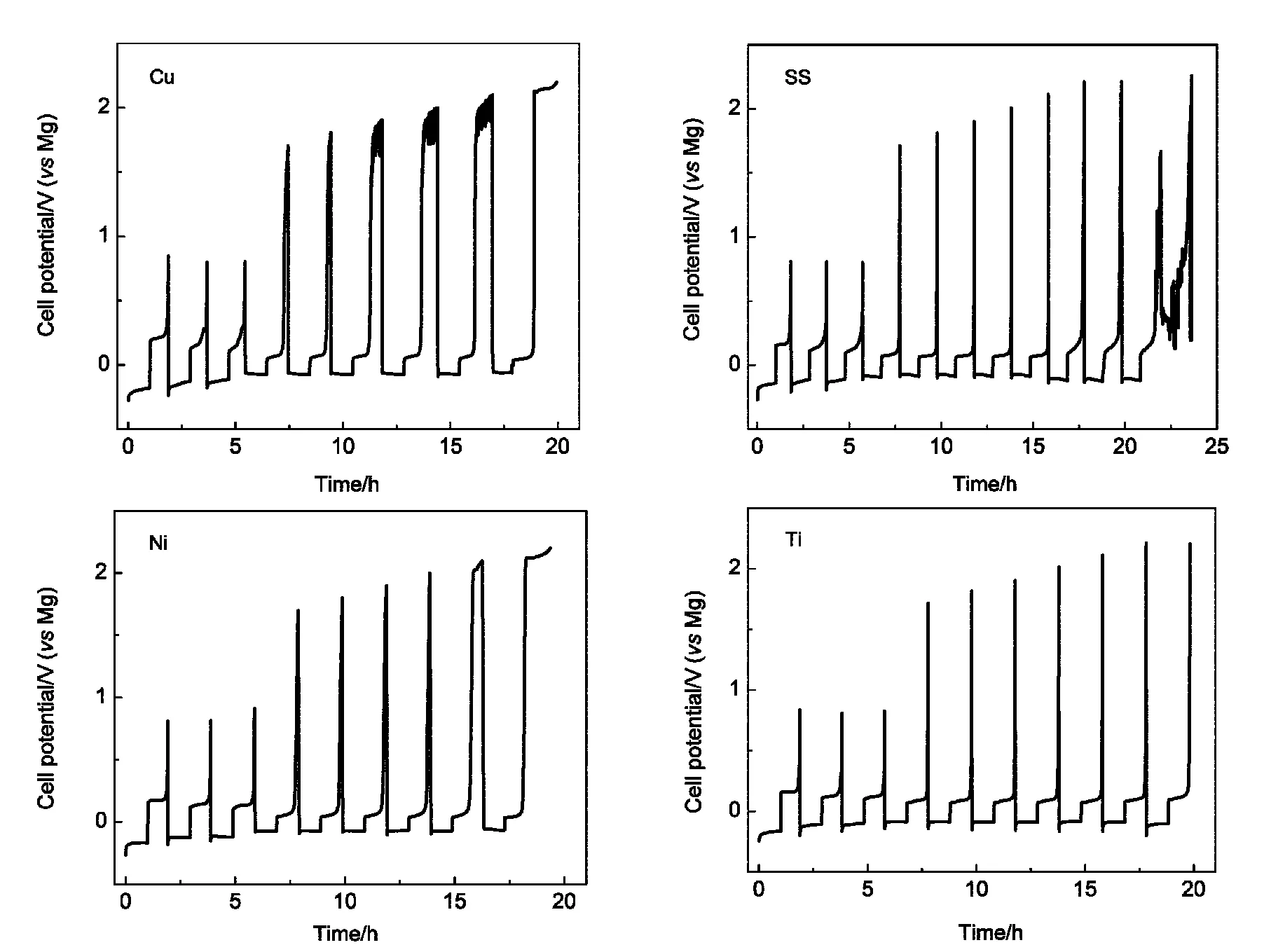
图9 不同集流体上镁沉积-溶出循环曲线Fig.9 Mg deposition-dissolution cycling curves on different current collectors
将充电截止电压分别设成负载了活性材料的集流体应该已发生腐蚀的充电截止电压(比图8中的较高,Cu:2.00 V,Ti:2.00 V,Ni:2.10 V,SS:2.10 V),测试完后同样将电池拆开,将集流体洗净干燥后进行SEM测试,结果如图10所示.可以看到没有负载活性材料的集流体除去钛之外也有腐蚀现象(图中圆圈标出处)出现,但并没有发生如图8中所示的明显的腐蚀现象.其中,钛集流体表面没有被腐蚀,而是被一层表面膜覆盖(图10(f)),保护其不受电解液的腐蚀.
以上结果表明实际电池体系中引起集流体腐蚀电位降低的原因是负载在集流体上添加了导电碳和粘结剂的正极材料涂覆层.集流体上涂覆层表面电流密度分布不均匀是造成这种现象的原因.当非完全均相的正极材料浆料涂覆在集流体表面时,其局部电导率大小差别较大.与高电导率接触的区域,流经正极材料/集流体界面的电流密度比与低电导率接触的区域高,当局部电流密度达到一个足够大的值时,此处的集流体将首先发生腐蚀.已发生腐蚀的位置更容易再次被腐蚀,引起凹点和周边区域剥落.

图10 镁沉积-溶出测试前后不同集流体表面的SEM形貌图Fig.10 Surface SEM images of different current collectors before and after magnesium deposition-dissolution measurements

图11 SS集流体上30%Super-P+70%PVDF/Mg扣式电池的放电-充电曲线Fig.11 Discharge-charge curve for 30%Super-P+70% PVDF/Mg coin cell with SS current collector PVDF:polyvinylidene fluoride

图12 30%Super-P+70%PVDF/Mg扣式电池放电-充电前(a)后(b)SS集流体除去Super-P和PVDF的表面SEM形貌图Fig.12 Surface SEM images of SS current collector removing Super-Pand PVDF for 30%Super-P+70% PVDF/Mg coin cell before(a)and after(b)dischargecharge measurement
为了进一步验证,我们选择充电截止电压较高的不锈钢作为集流体,将30%(质量分数)导电炭和70%的PVDF制成浆料涂覆在不锈钢上,制成极片后装配成扣式电池进行充放电测试,充电截止电压设为2.10 V.结果如图11所示,电池在充电过程中电压维持在1.96 V左右,一直充不上去.这个电压和前面负载了活性材料的不锈钢发生腐蚀的电压(1.93 V)相差不多.超声除去导电碳和粘结剂后集流体的SEM测试结果表明此时不锈钢也发生了腐蚀,如图12所示,这说明即使表面仅仅涂覆导电炭和PVDF也会促进电解液对集流体的腐蚀,降低集流体在电解液中发生腐蚀的电位.从图8中可以看出,这种作用对本身稳定性极好的钛集流体具有很强的破坏性,可能会打破其表面膜或阻碍表面膜的形成.表面膜的损害或消失改变了集流体/电解液的界面状况,使钛金属直接暴露在电解液中,使其在较低电位下也会发生腐蚀,稳定性能大大降低,导致Mo6S8在钛集流体上的循环性能最差(如图3所示).
4 结论
研究了不锈钢、镍、钛、铜集流体对Mo6S8/Mg可充镁电池电化学性能的影响.在不锈钢集流体上电池的极化是最小的,并且具有较好的循环稳定性;镍、铜次之;钛集流体上的极化最大,循环稳定性也最差.SEM测试表明(PhMgCl)2-AlCl3/THF“二代”电解液会对这些集流体造成腐蚀,四种集流体在电解液中的稳定性和正极材料在集流体表面状况的不同是造成电化学性能差异的主要原因.正极材料浆料对集流体的腐蚀也起到了促进作用,降低了集流体在电解液中的稳定性.
致谢:感谢福州大学测试中心黄清明老师在XRD图谱Rietveld全谱拟合方面给予的帮助.
(1) Aurbach,D.;Lu,Z.;Schechter,A.;Gofer,Y.;Gizbar,H.; Turgemann,R.,Cohen,Y.;Moshkovich,M.;Levi,E.Nature2000,407,724.doi:10.1038/35037553
(2) Saha,P.;Datta,M.K.;Velikokhatnyi,O.I.;Manivannan,A.; Alman,D.;Kumta,P.N.J.Prog.Mater.Sci.2014,66,1.doi: 10.1016/j.pmatsci.2014.04.001
(3) Kim,H.S.;Arthur,T.S.;Allred,G.D.;Zajicek,J.;Nweman,J. G.;Rodnyansky,A.E.;Oliver,A.G.;Boggess,W.C.; Muldoom,J.Nature Commun.2011,2,427.doi:10.1038/ ncomms1435
(4) Levi,E.;Gofer,Y.;Aurbach,D.Chem.Mater.2010,22, 860.doi:10.1021/cm9016497
(5) Shen,J.;Peng,B.;Tao,Z.L.;Chen,J.Prog.Chem.2010,22(2/3),515.[沈 健,彭 博,陶占良,陈 军.化学进展,2010,22(2/3),515.]
(6) Zheng,Y.P.;Nuli,Y.N.;Yang,J.;Chen,Q.;Wang J.L.Chem. Ind.Eng.Prog.2011,30(5),1025. [郑育培,努丽燕娜,杨军,陈 强,王久林.化工进展,2011,30(5),1025.]
(7) Zhao,Q.S.;Nuli,Y.N.;Guo,Y.S.;Yang,J.;Wang,J.L.Process.Chem.2011,23(8),1599.[赵青松,努丽燕娜,郭永胜,杨 军,王久林.化学进展,2011,23(8),1599.]
(8) Levi,M.D.;Lancry,E.;Gizbar,H.;Lu,Z.;Levi,E.;Gofer,Y.; Aurbach,D.J.Electrochem.Soc.2004,151(7),A1044.
(9) Chusid,O.;Gofer,Y.;Gizbar,H.;Vestfrid,Y.;Levi,E.; Aurbach,D.;Riech,I.Adv.Mater.2003,15(7-8),627.
(10) Aurbach,D.;Gizbar,H.;Schechter,A.;Chusid,O.;Gottlieb,H. E.;Gofer,Y.;Goldberg,I.J.Electrochem.Soc.2002,149(2), A115.
(11) Vestfried,Y.;Chusid,O.;Gofer,Y.;Aped,P.;Aurbach,D.Organomet.2007,26,3130.doi:10.1021/om061076s
(12) Viestfried,Y.;Levi,M.D.;Gofer,Y.;Aurbach,D.J.Electroana. Chem.2005,576,183.doi:10.1016/j.jelechem.2004.09.034
(13) Aurbach,D.;Suresh,G.S.;Levi,E.;Mitelman,A.;Mizrahi,O.; Chusid,O.;Brunelli,M.Adv.Mater.2007,19,4260.
(14) Mizrahi,O.;Amir,N.;Pollak,E.;Chusid,O.;Marks,V.; Gottlieb,H.;Larush,L.;Zinigrad,E.;Aurbach,D.J.Electrochem.Soc.2008,155(2),A103.
(15) Pour,N.;Gofer,Y.;Major,D.T.;Aurbach,D.J.Am.Chem.Soc.2011,133,6270.doi:10.1021/ja1098512
(16) Muldoon,J.;Bucur,C.B.;Oliver,A.G.;Sugimoto,T.;Matsui, M.;Kim,H.S.;Allred,G.D.;Zajicek,J.;Kotani,Y.Energy Environ.Sci.2012,5,5941.doi:10.1039/c2ee03029b
(17) Guo,Y.S.;Zhang,F.;Yang,J.;Wang,F.F.;Nuli,Y.N.;Hirano, S.I.Energy Environ.Sci.2012,5,9100.doi:10.1039/ c2ee22509c
(18) Wang,F.F.;Guo,Y.S.;Yang,J.;Nuli,Y.N.;Hirano,S.I.Chem.Commun.2012,48,10763.doi:10.1039/c2cc35857c
(19) Muldoon,J.;Bucur,C.B.;Oliver,A.G.;Zajicek,J.;Allred,G. D.;Boggess,W.C.Energy Environ.Sci.2013,6,482.doi: 10.1039/c2ee23686a
(20) Lv,D.P.;Xu,T.;Saha,P.;Datta,M.K.;Gordin,M.L.; Manivannan,A.;Kumta,P.N.;Wang,D.H.J.Electrochem. Soc.2013,160(2),A351.
(21) Yagi,S.;Tanaka,A.;Ichikawa,Y.;Ichitsubo,T.;Matsubara,E.J.Electrochem.Soc.2013,160(3),C83.
(22) Chen,Q.;Nuli,Y.N.;Yang,J.;Kailibinuer,K.;Wang,J.L.Acta Phys.-Chim.Sin.2012,28(11),2625.[陈 强,努丽燕娜,杨 军,凯丽比努尔·克日木,王久林.物理化学学报,2012, 28(11),2625.]doi:10.3866/PKU.WHXB201208032
(23) Lancry,E.;Levi,E.;Mitelman,A.;Malovany,S.;Aurbach,D.J.Solid State Chem.2006,179,1879.doi:10.1016/j. jssc.2006.02.032
(24) Lancry,E.;Levi,E.;Gofer,Y.;Levi,M.;Salitra,G.;Aurbach, D.Chem.Mater.2004,16,2832.doi:10.1021/cm034944+
(25) Peng,C.X.Research on the Compatibility between Current Collectors and New Ionic Liquid Electrolytes for Lithium Secondary Batteries.Ph.D.Dissertation,Shanghai Jiao Tong University,Shanghai,2008. [彭成信.锂离子电池集流体与新型离子液体电解液的相容性及界面电化学行为研究[D].上海:上海交通大学,2008.]
(26) Fu,L.Y.;Chen,Z.X.;Cai,L.K.;Zhu,H.F.;Zhou,H.Corrosion&Protection2000,21(7),294.[傅丽英,陈中兴,蔡兰坤,祝鸿范,周 浩.腐蚀与防护,2000,21(7),294.]
(27) Zhu,J.J.;Guo,Y.S.;Yang,J.;NuLi,Y.N.;Zhang,F.;Wang,J. L.;Hirano,S.I.J.Power Sources2014,248,690.doi:10.1016/ j.jpowsour.2013.09.124
(28) Mohtadi,R.;Matsui,M.;Arthur,T.S.;Hwang,S.J.Angew. Chem.Int.Edit.2012,51,9780.doi:10.1002/anie.201204913
(29) Shao,Y.Y.;Liu,T.B.;Li,G.S.;Gu,M.;Nie,Z.M.;Engelhard, M.;Xiao,J.;Lv,D.P.;Wang,C.M.;Zhang,J.G.;Liu,J.Scientific Reports2013,3,3130.
Effects of Cathode Current Collectors on the Electrochemical Performance of Rechargeable Magnesium Batteries
SU Shuo-Jian NULI Yan-Na*FEILURE Tuerxun YANG Jun WANG Jiu-Lin
(School of Chemistry and Chemical Engineering,Shanghai Jiao Tong University,Shanghai 200240,P.R.China)
The effects of different cathode current collectors(stainless steel,nickel,copper,and titanium)on the electrochemical performance of rechargeable magnesium batteries were investigated.Chevrel phase Mo6S8acted as the cathode,(PhMgCl)2-AlCl3/tetrahydrofuran(THF)(the second generation electrolyte)as the electrolyte,and magnesium as the anode.Constant current discharge-charge results indicated that the electrochemical polarization of the electrode on stainless steel was the smallest and its cycling stability was the best,followed by nickel,copper,and titanium.Microstructure analyses for the electrodes and current collectors before and after discharge-charge were analyzed to investigate the differing electrochemical performance.The electrolyte corroded the current collectors to differing degrees.The surfaces of the electrodes differed after coating the active material on different current collectors.The corrosion potentials of the current collectors decreased after loading the active material.This resulted in the current collectors being more susceptible to corrosion by the electrolyte.
Rechargeable magnesium battery;Cathode;Current collector; Electrochemical performance;Corrosion
The project was supported by the National Natural Science Foundation of China(21273147).
国家自然科学基金(21273147)资助项目
O646
10.3866/PKU.WHXB201411173www.whxb.pku.edu.cn
Received:September 24,2014;Revised:November 15,2014;Published on Web:November 17,2014.∗
