FC(O)SNCO的电子结构和光电离解离过程
2015-01-04赵春红佟胜睿葛茂发中国科学院化学研究所分子动态与稳态结构国家重点实验室北京分子科学国家实验室北京0090河北师范大学化学与材料科学学院石家庄05004
赵春红 佟胜睿 葛茂发 孙 政(中国科学院化学研究所,分子动态与稳态结构国家重点实验室,北京分子科学国家实验室,北京0090;河北师范大学化学与材料科学学院,石家庄05004)
FC(O)SNCO的电子结构和光电离解离过程
赵春红1,2佟胜睿1,*葛茂发1孙 政2,*
(1中国科学院化学研究所,分子动态与稳态结构国家重点实验室,北京分子科学国家实验室,北京100190;2河北师范大学化学与材料科学学院,石家庄050024)
由于拥有―C(O)S―和―NCO基团,FC(O)SNCO的分子和电子结构是非常有趣的.利用FC(O)SCl和AgNCO制备了FC(O)SNCO,并利用HeI光电子能谱(PES)、光电离质谱(PIMS)以及理论计算研究了其分子和电子结构.通过将实验、理论计算以及自然键轨道(NBO)分析结合起来,获得了FC(O)SNCO的最稳定分子构型.利用外壳层格林函数(OVGF)方法以及与相似化合物的比较,对其光电子能谱进行了指认.理论计算表明,对于中性分子最稳定的构型为syn-syn非平面构型,而电离后的离子最稳定构型为syn-syn平面构型.实验结果表明,第一电离能来自于S的孤对电子轨道,为10.33 eV.第二至第六电离能分别为12.03、13.23、13.77、14.78、15.99 eV,并对这些电离能进行了指认.在光电离质谱中产生了六个质谱峰,分别为SN+、FC(O)+、SNCO+、FC(O)SN+、C(O)SNCO+、FC(O)SNCO+•,其中FC(O)SNCO+•的峰是最强峰.结合HeI光电子能谱和理论计算,对PIMS进行了分析,并研究了可能的电离和解离过程并对其进行了讨论.
结构;HeI光电子能谱;光电离质谱;理论计算;电离和解离过程
1 Introduction
A wide series of studies on thiol ester compounds have been carried out because of their close relation to many biomolecules. For instance,acetyl CoA,is well known as a biological acetylating reagent,leading to a reductive scission of the S―S linkage of 6,8-thioctic acid to form a dithiol.1It is also of great importance in biochemistry as a condensing agent.Due to the important role against free radicals,the S-acyl-CoA and reduced glutathione GSH are also seen as important intermediates in the metabolism of acidic drugs.Some thiol esters also represent as latent medicine which can be reactivated through hydrolysis.2Needless to say,at present it is difficult to explain all the biological activity distinctly. Some significant aspects of them,however,may be examined by the investigation of simple model compounds,such as sulfenyl carbonyl compounds,XC(O)SY(X,Y:halogen).XC(O)SY compounds containing―C(O)S―are an interesting family of many theoretical and experimental investigations.Haas and Reinke3reported the preparation of the first such compound in 1966,namely ClC(O)SCl.Since then,several other members of this type of compounds have been characterized,e.g.,FC(O)SCl,4-7FC(O)SBr,8-10and ClC(O)SBr.10,11Recently,the X,Y in XC(O)SY represent not only halogen but also pseudohalogen and other substituent groups,e.g.,FC(O)SSCN,12CH3OC(O)SNCO,13FC(O)SOC(O)CH314and so on.The essentially planar nature of the XC(O)SY skeleton gives rise to syn and anti conformational options.Gas electron diffraction,4vibrational analysis,5and calculations6demonstrated that FC(O)SCl exists as a mixture of two planar forms with the energy of anti conformation(C=O bond anti with respect to the S―Cl bond)being higher than that of the syn conformer.In all the reported cases,the syn conformation represents the most stable structure.8,10,15
Isocyanates as one of the pseudohalide derivatives are important precursors in the synthesis of polymer materials.16The production is based on a nucleophilic addition reaction which takes place via the nucleophile's attack on the carbon of the NCO group.The synthetic procedure gives high yields and exhibits no side reactions.Pseudohalides are examples of distorted linear triatomic systems.It was difficult to predict the conformational stability of simple isocyanates and may also be different in different aggregation states.Several studies have been carried out on the conformational and spectroscopic properties of organoisocyanate molecules.For example,in methoxycarbonylsulfenyl isocyanate,CH3OC(O)SNCO,13the C=O double bond synperiplanar with respect to the S―N single bond was the most stable form,which confirmed by Fourier transform infrared (FTIR)spectroscopy,ultraviolet visible(UV-Vis)spectroscopy,X-ray diffraction(XRD),Raman spectroscopy,and theoretical calculations.Gas-phase electron diffraction(GED),microwave, calculations,and vibrational analysis were used to study the conformational and structural properties of acetylisocyanate CH3C(O)NCO,17all results predicted that in the gas phase and ambient temperature the cis conformer(isocyanate group cis to the carbonyl bond)was more stable than the trans conformer. The studies on conformational properties of CCl3C(O)NCO were close to those of CH3C(O)NCO.18In the liquid phases the cis conformer was more stable than the trans conformer indicated by Raman and IR studies,while only the cis conformer was present for CF3C(O)NCO in the solid phase.19
(Fluorocarbonyl)sulfenyl isocyanate,FC(O)SNCO,is a very meaningful molecule,which is one of the examples of the XC(O)SY compounds including isocyanate(NCO)group.The conformation of the gaseous FC(O)SNCO has been studied by GED,FTIR,and quantum chemical calculation.20In addition,the vibrational analysis and matrix-photochemical study have been reported.21However,the electronic structure of FC(O)SNCO has never been studied.Our group has a long-term concern with the generation and electronic structures of unstable molecules.22,23The HeI photoelectron spectroscopy(PES),together with theoretical calculation will play an important role in studying electronic structures of compounds.Comparing with the structure investigations,the electronic structures of these unstable molecules can be studied.In this work,the HeI photoelectron spectrum and photoionization mass spectrum(PIMS)of FC(O)SNCO were first reported.The electronic structure of the compound was analyzed based on the HeI photoelectron spectrum in combining with theoretical calculation and photoionization mass spectroscopy.
2 Experimental
2.1 Sample preparation
In the preparation of isocyanates,silver isocyanate (AgOCN,Alfa,>97%)was found to be a perfect precursor.24In this work,FC(O)SNCO was prepared by substitution reaction of FC(O)SCl and AgOCN at-80°C for 12 h according to literature.25The reaction route was illustrated as below:

The reactant FC(O)SCl was prepared by ClC(O)SCl(97%,Alfa Aesar)and TlF(99%,Acrod,USA)referring to the previous report,26and its potential energy(PE)and IR spectra were identical to the reports previously.27AgOCN was all purchased from Alfa Aesar Company.Before reaction,AgOCN was dried for 2 h in vacuum(1.33×10-6Pa).The IR spectrum of gaseous FC(O)SNCO was also obtained.Compared with the reported IR spectroscopy,it was verified that the synthetic compound was FC(O)SNCO.21
2.2 Instruments
A double-chamber UPS-II instrument was used to record the photoelectron spectrum as reported before.28,29The instrument was specially designed to detect unstable species.Alittle ofAr gas and CH3I were acceded to the sample flow tube to calibrate the experimental vertical ionization potentials during the experiments.The spectral resolution of HeI spectrum was about 30 meV.The mass spectrum was recorded by a time-of-flight mass spectrometer and mounted directly to the double-chamber UPS-II instrument.The soft ionization was supplied by single wavelength HeI radiation. The instrument was represented in detail in the previous paper.29Under identical conditions the photoelectron and photoionization mass spectra can be recorded within seconds of each other.
2.3 Theoretical computation
The geometry of the neutral ground state was optimized using MP2/6-311++G(d,p)and B3LYP/6-311++G(d,p).Harmonic vibrational frequencies were also calculated to check whether the optimized structure was a stationary point or a saddle point.The potential functions for internal rotation around the C―S were performed to search for other possible formers of FC(O)SNCO at the MP2/6-311++G(d,p),B3LYP/6-311++G(d,p),and HF/6-311++ G(d,p)levels.At the B3LYP/6-311++G(3df,3pd)level,the natural bond orbital(NBO)calculations were calculated.To assign the photoelectron spectrum,the out-valence Green's function(OVGF) calculation,which included sophisticated correlation effects of the self-energy,was applied to the most stable conformers of FC(O)SNCO to give accurate results of the vertical ionization energies.Gaussian 03 program was used for calculations.30Threedimensional molecular orbital(MO)plots were obtained with the Gauss View program,and each orbital displayed with the 0.06 isodensity value was oriented in a way that allowed for the best view.The Mulliken population analysis was applied to assign the charges for both neutral and cationic-radical forms.
3 Results and discussion
3.1 Molecular structure
In order to seek other possible conformers of FC(O)SNCO,the potential functions for internal rotation around the C―S were performed by rotating the torsional dihedral angle(δFCSN)in steps of 10°ranging from 0°to 360°at the MP2/6-311++G(d,p), B3LYP/6-311++G(d,p),and HF/6-311++G(d,p)levels(Fig.1).All these methods predicted that the syn-syn structure(δFCSN=180°) was the global minimum form.At the same time,a second minimum was anti-anti structure(δFCSN=0°).The structural optimization and vibrational analysis indicated that the syn-syn and antianti conformers of FC(O)SNCO were stable,which were shown in Fig.2.Relative Gibbs free energies(ΔG)for the two stable conformers of FC(O)SNCO at 298 K after optimization were calculated.The second stable form was anti-anti former with the energy gaps of 8.00,7.29,and 10.84 kJ·mol-1based at the HF/6-311++G(d,p),B3LYP/6-311++G(d,p),and MP2/6-311++G(d,p) levels,respectively.All the computational methods predicted that the syn-syn conformer was the most stable.This can be explained by the sulfur lone-pair and steric interactions between FC(O)and NCO groups.

Fig.1 Relative energies of FC(O)SNCO with different dihedral angles(δFCSN)calculated at the HF/6-311++G(d,p),B3LYP/6-311++ G(d,p),and MP2/6-311++G(d,p)levels

Fig.2 Schematic representation of FC(O)SNCO
Probably the most interesting and contrasting results uncovered by the potential energy curves shown in Fig.1 are the very different rotational barriers obtained for the two studied compounds. Computed energy barriers Eafor the syn→anti internal rotation were derived from fully optimized transition state(TS)structures at B3LYP levels of theory.The TS(Fig.3)structures were verified by an intrinsic reaction coordinate calculation to connect the conformers along the reaction path for the normal coordinate of the imaginary frequency.
To gain some deep information on the stabilities of syn-syn and anti-anti conformations of FC(O)SNCO,NBO calculations were also performed on these two conformers.The most important orbital interactions occur between the two sulfur lone pairs relevant to the conformational properties,lpπS(2)and lpσS(1)[lp: lone pair;the lpσS(1)orbital is parallel to the σ*(C1―O1)bond,the lpπS(2)orbital is parallel to the π(C1=O1)bond].The occupancies of lpσS(1)and lpπS(2)are 1.98e and 1.79e,respectively.The lower occupancy of lpπS(2)indicates its strong electron-donor capacity. The strongest interactions are due to donations from lpπS(2)to theunoccupied molecular orbits π*(C1=O1)[lpπS(2)→π*(C1=O1)] and π*(N=C2)[lpπS(2)→π*(N=C2)].This is evidenced by high occupancy of the two antibonding orbitals π*(C1=O1)(0.27e), anti-anti form,which contributes to stabilization of an approximate planar structure for FC(O)SNCO by resonance(mesomeric effect).As shown in Table 1,these interactions contribute similarly to stabilization energies for both the syn-syn and anti-anti forms.However,delocalization of lpσS reveals a strong conformational dependence.In account with the anomeric effect,the lpσS(1)→σ*(C1―O1)interaction stabilized the syn-syn form by 21.87 kJ·mol-1,while the corresponding delocalization in the antianti form is below the threshold for printing(2.09 kJ·mol-1).On the other hand,the syn-syn form is favored through lpσS(1)→σ*(C1―F)delocalization by 11.77 kJ·mol-1.Thus,considering the electronic contributions of both lone pairs at the sulfur atom,the syn-syn form is favored by 22.73 kJ·mol-1.Similar results were reported also for other sulfenylcarbonyl molecules.31

Fig.3 Optimized structures,labeling of the atoms,and relative energies of the syn,anti conformations,and rotational transition state(TS)of FC(O)SNCO molecule at the B3LYP/6-311++G(d,p)level

Table 1 NBO stabilization energies for orbital interactions associated with sulfur lone pair(lp)in FC(O)SNCO calculated at the B3LYP/6-311++G(3df,3pd)level
In the previous work,the conformational properties of FC(O)SNCO have been investigated by gas electron diffraction,20theoretical calculation,and matrix infrared spectra.21Only a synsyn conformation(carbonyl C=O syn with S―N and the NCO group syn to C―S)was confirmed to be the most stable conformer at ambient temperature.The result of our theoretical calculation agreed with the previous studies.Therefore,our calculations were all based on the syn-syn conformer.
Most of the series of compound used B3LYP method,such as CH2=CHC(O)NCO,32FC(O)SOC(O)CH3,14CH3OC(O)SCH3,13FCOSSCN,12FC(O)SCN,31and so on.For neutral and cationic FC(O)SNCO,full geometry optimizations and frequency calculations were performed on the syn-syn conformer at the B3LYP/ 6-311++G(d,p)basis set.Table 2 compared experimental and calculation geometric parameter of FC(O)SNCO.The structure obtained by theoretical calculations was in accordance with the GED results.20Fig.2 showed the geometric parameters of the ground cationic-radical form FC(O)SNCO+•,which were also calculated by the B3LYP approach at the same basis set.After the vibrational analysis,the most stable conformer of the cationicradical form was Cssymmetry(Fig.2).

Table 2 Experimental and calculated geometric parameters for syn-syn conformer of FC(O)SNCOa
3.2 Photoelectron spectroscopy
In combination with theoretical calculations,PES with HeI resonance source(58.4 nm)is an effective method to investigate the electronic structure of unstable compounds and free radicals.33By HeI photoelectron energy analysis,the valence shell structure of the molecule can be obtained,especially for investigating similar molecules.34The HeI photoelectron spectroscopy spectrum of FC(O)SNCO is presented in Fig.4.Table 3 listed the experimental ionization potentials(IP in eV)and theoretical vertical ionization energies(Evin eV)as well as corresponding characters of each occupied molecular orbital.The OVGF pole strengths in Table 4 are all larger than 0.85,which agrees with a one-electron depiction of ionization.If the OVGF pole strengths are smaller than 0.85,OVGF approach would be unapplicable because a severe breakdown of the orbital(or one-electron)picture of ionization should be expected.35To obtain the ionization energies for FC(O)SNCO,OVGF calculations were carried out before assigning the spectrum.All the calculations were based on themost stable conformer syn-syn.Fig.5 shows the characters of the some occupied molecular orbitals of FC(O)SNCO.
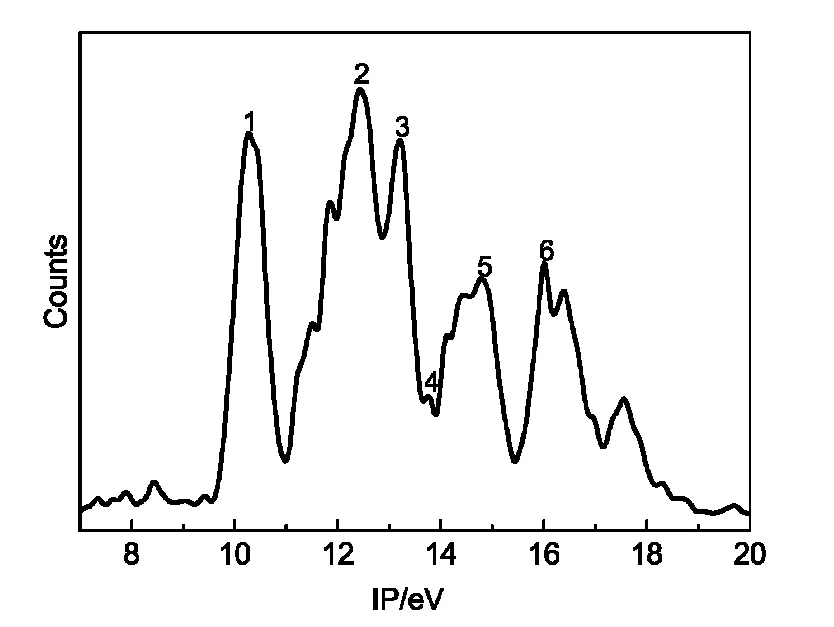
Fig.4 Photoelectron spectroscopy of FC(O)SNCO
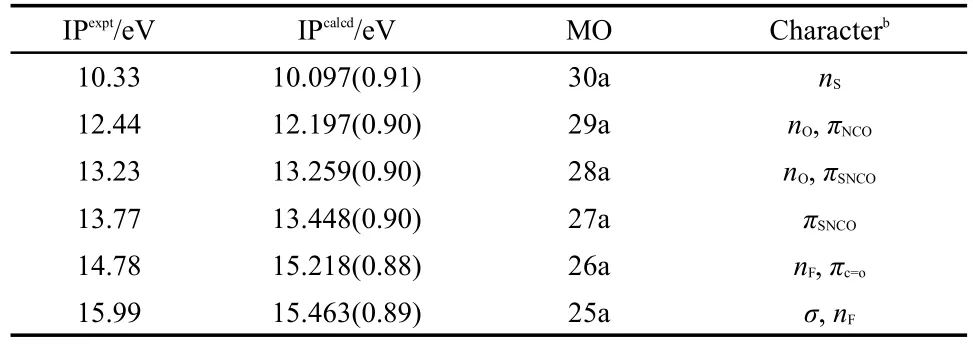
Table 3 Experimental vertical ionization potentials(IPexpt), calculated vertical ionization potentialsa(IPcalcd)as obtained by OVGF/6-311++G**and MO characters for FC(O)SNCO
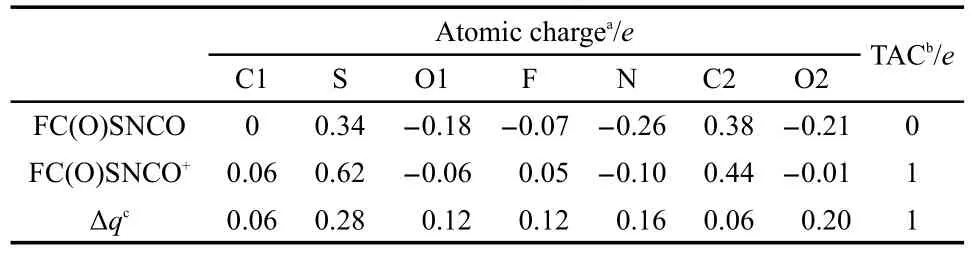
Table 4 Atomic charges for the neutral(syn-syn)and cationicradical(syn-syn)conformer of FC(O)SNCO as obtained by the B3LYP/6-311++G(d,p)level
It can be clearly seen from Table 3,the ionization energies of different bands are in consistent with the calculated values of OVGF method.Combining with band profiles,relative band intensities,comparison with the similar compounds,and theoretical method(OVGF calculation),the bands in the photoelectron spectra can be reliable assignments.The assignment of the photoelectron spectrum of FC(O)SNCO is obtained easily,because lone pairs generally display no fine structure.
In the photoelectron spectrum,the first ionization band of FC(O)SNCO was at 10.33 eV that can be contributed to the ionization process from the highest occupied molecular orbital(HOMO)[nπ(S)],which can be direct-viewing as a lone pair formally localized on the sulfur atom.This is similar to FC(O)SCl,its precursor.36The first ionization energy(10.7 eV)of FC(O)SCl also is from the ionization of lone pair of the sulfur atom.For FC(O)SNCO the first vertical ionization energy is slightly lower(0.37 eV)than FC(O)SCl in photoelectron spectrum.The reason was that the highly polar S+―O-of the S―O bond in sulfenic esters RSOR'.Because the electronegativity of F is larger than that of CH3O,it results in more electrons on S lone pair of FC(O)SNCO.The first ionization of FC(O)SNCO is higher (0.77 eV)than that of CH3OC(O)SNCO(9.56 eV).13The first band of ClC(O)SCl37is located at 10.36 eV,which can be assigned to the removal of an electron from the sulfur lone pair,the highest occupied molecular orbital(HOMO).
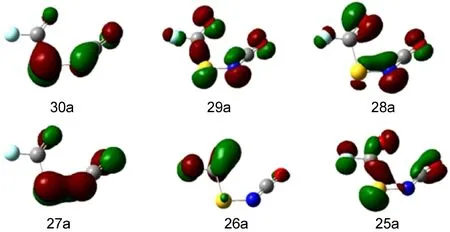
Fig.5 Characters of the occupied molecular orbitals (MOs)for FC(O)SNCO
Fig.6 shows a diagrammatic representation of the experimentally determined IPs for some molecules formally containing the sulfenylcarbonyl group,13,36-40which can also be known as―C(O)S―containing compounds.Taking CH3OC(O)SNCO13as a reference,it can be observed a reduction of the IP values because of the substitution of the NCO by electron-releasing alkyl groups.On the other hand,substitution of the NCO or SCN groups by more electronegative groups can lead to higher IPvalues.
The―C(O)S―compounds previously studied had the similar first ionization potential values,which can be assigned as a lone pair formally localized on the sulfur atom(Fig.6).For instance, the ionization potential values of CH3OC(O)SY series,ClC(O)SY series and FC(O)SY series,the strong inductive effect of Cl atom or F atom can raise the ionization energy of sulfur lone pair orbital nS,with respect to the CH3OC(O)SY molecules.The electronegativity of F is the strongest,and CH3is the weakest.So the ionization potentials of FC(O)SY compounds are higher thanthose of CH3C(O)SY compounds,and ClC(O)SY compounds are in the middle.Meanwhile,the electronegativity of―NCO is between―CH3and―SCN.So the first ionization energy of FC(O)SNCO at 10.33 eV between those of CH3OC(O)SNCO13and FC(O)SSCN12is very reasonable.

Fig.6 Correlation diagram of the IPs of―C(O)S―containing compounds
However,this effect must be interpreted in terms of delocalization effects,rather than as inductive effects of the substituents. This stabilization seems to be caused by the planar XC(O)S moiety around the C(sp2)and is independent of the nature of X. The first IP values of two model compounds CH3OC(O)SCH338and ClC(O)SCH339are 9.46 and 10.11 eV,respectively.
In order to analyze the nature of the cation formed in the first ionization process,further calculations(B3LYP/6-311+G(d,p)) were performed.It demonstrated that the atomic charges were a palpable fraction localized at sulfur atoms,with delocalized all over the molecule(Table 4).Table 3 summarized the optimized structural parameters of the cationic-radical form.The adiabatic potential of FC(O)SNCO is 9.87 eV,which was derived from the different energies between the neutral and ground cationic-radical forms at B3LYP/6-311G(d,p)level of theory.Further vibrational calculations were performed for the cationic-radical form,which resulted in shift(128 cm-1)of the wavenumber for νC=Otoward higher values,as compared to the neutral molecular,while the νC―Swavenumber exhibits a shift of 23 cm-1in the opposite direction.With the interruption of the mesomeric effect and a reinforced bond character in the C=O bond and decreasing in the corresponding S―C bond,the picture of an electron ionization mainly localized at the S atom.
The second band of FC(O)SNCO at 12.44 eV in the photoelectron spectrum,which is due to the electrons ionization from the orbital 29a(Fig.5),can be assigned to the oxygen lone pair noof the carbonyl group with some admixture out-of-plane nonbonding orbital on the NCO moiety.This value is consistent with the IPs of the oxygen lone pair orbitals in the correlative species. The second band of FC(O)SCl35is at 12.1 eV which can be contribute to the removal of an electron from the lone pair electron of oxygen atom of the carbonyl group,and is higher than that of FC(O)SNCO by 0.34 eV.The forth IP of FC(O)SSCN12at 12.73 eV is mainly due to the lone pair electron of oxygen atom.In contrast to FC(O)SCl and FC(O)SSCN,the third ionization band of FC(O)SC(O)CH341is at 11.80 eV(nO(FC(O))).The trend showed a clear dependence on the substituents attached to the sulfur atom. Therefore,in the series of FC(O)SX(X=NCO,SCN,C(O)CH3) species,the FC(O)SNCO molecule possessed higher stabilization of the oxygen lone pair.
The bands at 13.23 and 13.77 eV can be assigned of ionization electrons on the in-plane lone pair of carbonyl oxygen π'COand the in-plane π'SNCO.The orbit π'SNCOconsists the the in-plane of isocyanate and in-plane lone pair of sulfur.The fifth band originates from ionizations related to the FC(O)moiety.Interactions between the πC=Oand nFlocalized orbitals become effective in the planar geometry of the FC(O)moiety.Howere,it is impossible to have this interaction in the in-plane lone-pair orbital.From the above, the band at 14.78 eV may involve πC=Oand nForbitals.The related species had the similar ionization potentials of the C=O group orbitals,such as CH3OC(O)SCH338(14.18 eV),FC(O)SC(O)CH341(14.10 eV),FC(O)SCl36(14.9 eV),FC(O)SSCH336(14.8 eV)and FC(O)SSCN12(14.17 eV).The πC=Oorbital is destabilized by the electron donating effect and stabilized by the resonance interaction with the fluorine non-bonding orbital.
The sixth band at 15.99 eV is just ascribed to the ionization of the orbital σ and nF.The band at 15.36 eV of FCOSSCN is mainly due to the lone pair electron of fluorine atom.The corresponding bands inpropynoyl fluoride and formyl fluoride were at 15.4 and 15.5 eV,12respectively,which exhibited remarkably similar shapes. The band at higher ionization region(>16.00 eV)can be attributed to the ionization of σ orbital,and can be attributed to the ionization of inner orbital of FC(O)SNCO,which may be difficult to assign certainly.
3.3 Photoelectron mass spectrometry
The PIMS spectroscopy is depicted in Fig.7.Combining the PES and the theoretical results,the PIMS results can be analyzed. The assignments mainly depended on the mass number of the fragments.It shows six peaks:SN+(m/z=46),FC(O)+(m/z=47), FCOSN+(m/z=93),C(O)SNCO+(m/z=102),SCNO+(m/z=74)and the parent molecular ion M+•,among which the dominant features was the parent molecular ion M+•peaks.

Fig.7 PIMS spectrum of FC(O)SNCO
The energies of the identified fragments and the adiabatic ionization of FC(O)SNCO were computed,to confirm the level of electronic structure theory which is used in the energetics calculation of dissociation pathways.Table 5 shows the experimentally measured values and the calculated results.The experimental ionization energy data of the FC(O)S,SNCO,C(O)SNCO, and FC(O)SN radicals are not available.Since the experimental ionization energies are in very good agreement with the calculated ionization energies of the other fragments,the calculated ionization energies for these radicals can be used to estimate the actual values.Theoretical calculation was performed to estimate the energies of the dissociation pathway.These ionization energies are necessary to construct the energy diagrams of different pathways for the ionic dissociation of the parent ion shown in Fig.8.
3.4 Dissociation of fluorocarbonyl sulfenyl isocyanate
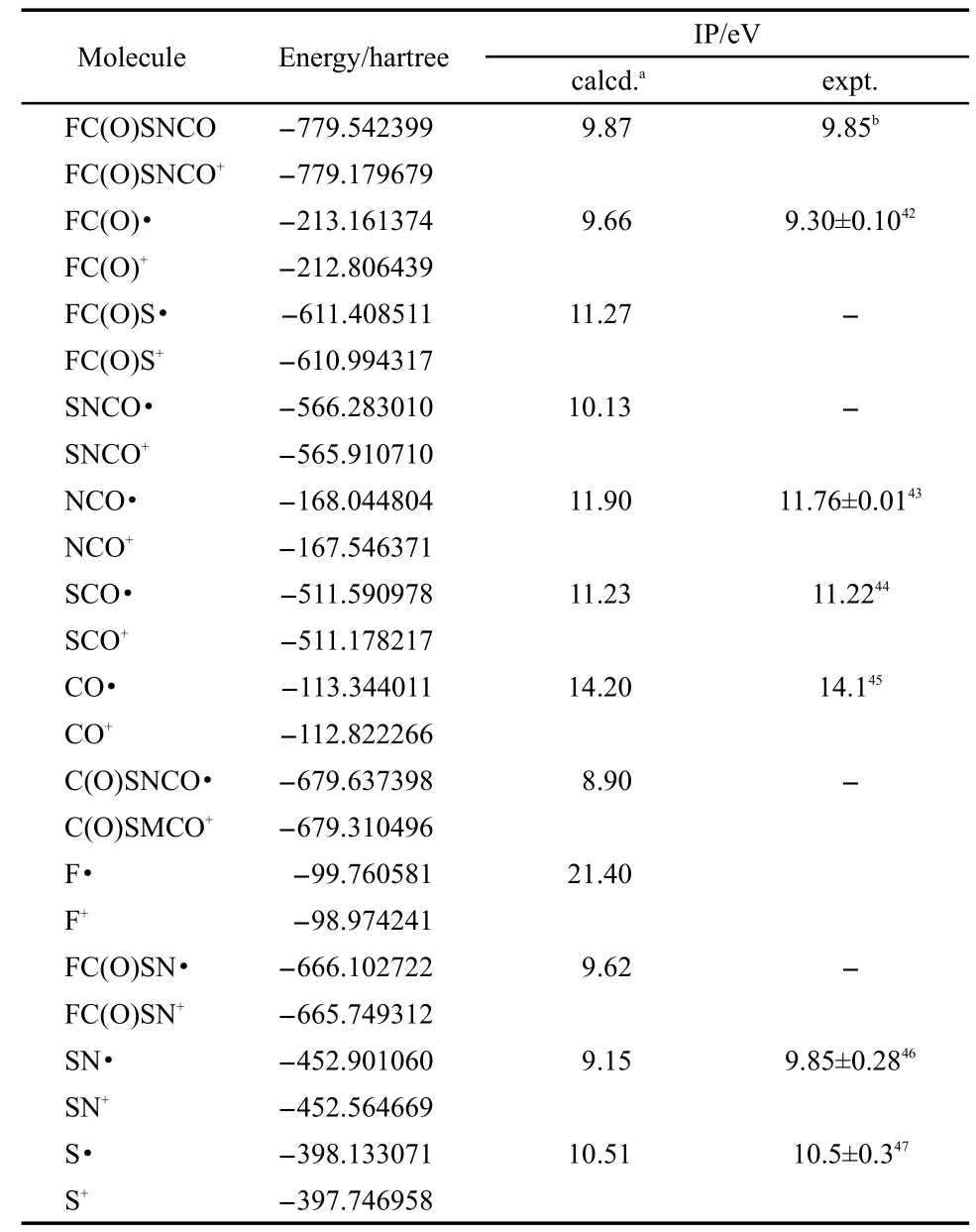
Table 5 Calculated and experimental ionization potentials of FC(O)SNCO and its fragments
Besides the studies of the PES and PIMS,the dissociation processes of FC(O)SNCO were also investigated in this work.
The neutral ground state of FC(O)SNCO possesses the C1symmetry with a non-planar conformer(δCSNC=-22.33°).But FC(O)SNCO+•has an interesting and important feature with Cssymmetry,with F and SNCO synperiplanar with C=O.In other word,FC(O)SNCO is a planar conformer(δCSNC=0°).Removing one electron from the HOMO,lone pair of sulfur atom can influence the geometric parameters.The change in bond lengths is obvious:the rC1―Sbond elongates to 0.1907 nm.The occupancy of C1―S band also reduces from 1.97e(electron charge)(neutral) to 0.98e(cation).Therefore,these changes in the geometric parameters can strongly influence the removal of the σC1―Sbonding electrons.The strongest interaction describing a conjugation (mesomeric effect)between the lpπS lone pair and the π*(C1―O) bond[lpπS(2)→π*(C1―O)]is disappeared.All these changes can weaken the C1―S band.All these results demonstrate that the parent ion is easy to have a dissociation on the C1―S single bond to form the FC(O)+/SNCO·pair or the SNCO+/FC(O)·pair.
The possible dissociation pathways of FC(O)SNCO+•are shown in Fig.8.The dissociation of FC(O)SNCO+•into FC(O)+/ SNCO·(2.46 eV)and SNCO+/FC(O)·(2.93 eV)are energetically favorable over most ways.However,the dissociation of FC(O)SNCO+•into FCOSN+/CO·(2.35 eV)is the easiest channel,which is lower than FC(O)+/SNCO·by 0.11 eV.The other two dissociation pathways are SNCO+/FC(O)·(2.93 eV) and C(O)SNCO+/F·(2.96 eV).Through the above four ways,we can obtain FC(O)SN+,FC(O)+,SNCO+,and C(O)SNCO+.Because SNCO+can easily dissociate into SN+/CO·,FC(O)SN+into SN+/ FC(O)·,and F·/C(O)SNCO+into F·/2CO·/SN+,which only need 0.05,0.63,and 1.57 eV,respectively.This is why SN+appeared in photoionization mass spectrum and had the second highest intensity.The energies of the dissociation into FC(O)+,SNCO+, FC(O)SN+,C(O)SNCO+are very nearly,so the intensities of their bands are similar.Thus,the ion intensities observed in the photoionization mass spectrum can be explained by anticipating the dissociation pathways.

Fig.8 Schematic energy profile(eV)showing ionization and dissociation pathways of FC(O)SNCO calculated at the B3LYP/6-311++G(d,p)level
4 Conclusions
FC(O)SNCO was synthesized by the reaction between AgOCN and FC(O)SCl.The structural and electronic properties of FC(O)SNCO were studied by combination of experiment,theoretical studies,and NBO analysis,before characterizing by PES and PIMS.The result showed that the most stable conformer for the title molecule was the syn-syn from with C1symmetry.After ionization,the cationic-radical FC(O)SNCO+•adopted syn-syn structure with Cssymmetry and planar conformer.Combining with OVGF calculation,the PES spectrum was assigned.Because of the geometric change after ionization,the first vertical ionization energy(10.33 eV)was different from the first adiabatic ionization energy(9.87 eV).In the PIMS of FC(O)SNCO,there were six peaks:SN+,FC(O)+,SNCO+,FC(O)SN+,C(O)SNCO+, and FC(O)SNCO+•.According to the calculated bond dissociation energies,the investigations of possible ionization and dissociation processes were presented.Our investigation on the electronic structure of fluorocarbonyl sulfenyl isocyanate may have further implications for the understanding the chemical behavior of FC(O)SNCO.
(1) Watson,J.D.;Gann,A.;Baker,T.A.;Levine,M.;Bell,S.P.; Losick,R.;Harrison,S.C.Molecular Biology of the Gene,7th ed.;CSH Press:New York,2013;pp 66-67.
(2) Ibarra,C.A.;Chowdhury,P.;Petrich,J.W.;Atkins,W.M.J.Biol.Chem.2003,278,19257.doi:10.1074/jbc.M301566200
(3) Hass,A.;Reinke,H.Angew.Chem.Int.Edit.1967,6,705.
(4) Della Vedova,C.O.;Varetti,E.L.;Aymonino,P.J.Can.J. Spectrosc.1983,28,107.
(5) Della Vedova,C.O.;Cutin,E.H.;Jubert,A.H.;Varetti,E.L.; Aymonino,P.J.Can.J.Spectrosc.1984,29,130.
(6) Mack,H.G.;Oberhammer,H.;Della Vedova,C.O.J.Phys. Chem.1991,95,4238.doi:10.1021/j100164a014
(7) Romano,R.M.;Della Vedova,C.O.;Boese,R.J.Mol.Struct.1999,475,1.doi:10.1016/S0022-2860(98)00439-6
(8) Della Vedova,C.O.J.Roman Spectrosc.1989,20,729.doi: 10.1002/jrs.v20:11
(9) Della Vedova,C.O.;Mack,H.G.Inorg.Chem.1993,32,948. doi:10.1021/ic00058a032
(10) Della Vedova,C.O.;Hass,A.Z.Anorg.Allg.Chem.1991,600, 145.
(11) Della Vedova,C.O.Spectrochim.Acta1990,46A,1073.
(12) Tong,S.R.;Du,L.;Yao,L.;Ge,M.F.;Della Vedova,C.O.Eur. J.Inorg.Chem.2008,3987.
(13) Ma,C.P.;Ge,M.F.J.Mol.Struct.2008,892,68.doi:10.1016/j. molstruc.2008.04.061
(14) Tong,S.R.;Ge,M.F.;Wang,W.G.;Della Vedova,C.O.J.Mol.Struct.2009,921,274.doi:10.1016/j. molstruc.2009.01.005
(15) Della Vedova,C.O.;Varetti,E.L.;Aymonino,P.J.Can.J. Spectrosc.1984,29,69.
(16) Ulrich,H.Chemistry and Technology of Isocyanates;Wiley: New York,1996.
(17) Mack,H.G.;Oberhammer,H.;Della Vedova,C.O.J.Mol. Struct.1992,265,359.doi:10.1016/0022-2860(92)80113-V
(18) Badawi,H.M.;Forner,W.;Abu-Sharkh,B.F.;Oloriegbe,Y.S.J.Mol.Model.2002,8,44.doi:10.1007/s00894-001-0066-5
(19) Durig,J.R.;Guirgis,G.A.;Krutules,K.A.J.Mol.Struct.1994,328,97.doi:10.1016/0022-2860(94)08388-6
(20) Gobbato,K.I.;Ulic,S.E.;Della Vedova,C.O.;Mack,H.G.; Oberhammer,H.Chem.Phys.Lett.1997,266,527.doi: 10.1016/S0009-2614(97)00040-7
(21) Ulica,S.E.;Hermannc,A.;Della Vedova,C.O.J.Mol.Struct.2002,641,233.
(22) Ge,M.F.;Wang,J.;Zhu,X.J.;Sun,Z.;Wang,D.X.J.Chem. Phys.2000,113,1866.doi:10.1063/1.481990
(23) Cao,X.Y.;Wu,W.;Wang,D.;Ge,M.F.;Wang,D.X.Acta Phys.-Chim.Sin.2000,16,491.[曹晓燕,吴 伟,王 东,葛茂发,王殿勋.物理化学学报,2000,16,491.]doi:10.3866/ PKU.WHXB20000603
(24) Yao,L.;Zeng,X.Q.;Ge,M.F.;Wang,W.G.;Sun,Z.;Du,L.; Wang,D.X.Eur.J.Inorg.Chem.2006,2006,2469.
(25) Erben,M.F.;Della Védova,C.O.;Willner,H.;Trautner,F.; Oberhammer,H.;Boese,R.Inorg.Chem.2005,44,7070.
(26) Erben,M.F.;Romano,R.M.;Della Védova,C.O.J.Phys. Chem.A2004,108,3938.doi:10.1021/jp038058y
(27) Haas,A.;Reinke,H.Chem.Ber.1969,102,2718.
(28) Du,L.;Yao,L.;Ge,M.F.J.Mol.Struct.2008,882,146.doi: 10.1016/j.molstruc.2007.09.024
(29) Du,L.;Yao,L.;Ge,M.F.J.Phys.Chem.A2007,111, 11787.doi:10.1021/jp075164h
(30) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision B.03;Gaussian Inc.:Pittsburgh,PA,2003.
(31) Ramos,L.A.;Ulic,S.E.;Romano,R.M.;Erben,M.F.; Lehmann,C.W.;Bernhardt,E.;Beckers,H.;Willner,H.;Della Vedova,C.O.Inorg.Chem.2010,49,11142.doi:10.1021/ ic101741e
(32) Ge,M.F.;Ma,C.P.;Xue,W.J.Phys.Chem.A2009,113, 3108.doi:10.1021/jp8110277
(33) Du,L.;Yao,L.;Zeng,X.Q.;Ge,M.F.J.Phys.Chem.A2007,111,4944.doi:10.1021/jp070601d
(34) Zeng,X.Q.;Ge,M.F.;Du,L.;Sun,Z.;Wang,D.X.J.Mol. Struct.2006,800,62.doi:10.1016/j.molstruc.2006.03.080
(35) Deleuze,M.S.;Giuffreda,M.G.;François,J.P.J.Phys.Chem. A2002,106,5626.doi:10.1021/jp014260u
(36) Erben,M.F.;Della Védova,C.O.Inorg.Chem.2002,41, 3740.doi:10.1021/ic020046o
(37) Gerones,M.;Erben,M.F.;Romano,R.M.;Della Vedova,C. O.;Yao,L.;Ge,M.F.J.Phys.Chem.A2008,112,2228.doi: 10.1021/jp7101034
(38) Ma,C.P.;Ge,M.F.J.Mol.Struct.2008,891,221.doi:10.1016/ j.molstruc.2008.03.030
(39) Gerones,M.;Erben,M.F.;Ge,M.F.;Cavasso,R.L.;Romano, R.M.;Della Vedova,C.O.J.Phys.Chem.A2010,114, 8049.doi:10.1021/jp102179w
(40) Du,L.;Yao,L.;Ge,M.F.Eur.J.Inorg.Chem.2007,2007, 4514.
(41) Tong,S.R.;Ge,M.F.;Wang,W.G.;Della Vedova,C.O.J.Mol.Struct.2009,919,83.doi:10.1016/j.molstruc. 2008.08.017
(42) Buckley,T.J.;Johnson,R.D.;Huie,R.E.;Zhang,Z.;Kuo,S. C.;Klemm,R.B.J.Phys.Chem.1995,99,4879.doi:10.1021/ j100014a002
(43) Dyke,J.M.;Jonathan,N.;Lewis,A.E.;Mills,J.D.;Morris,A.J.Mol.Phys.1983,50,77.doi:10.1080/00268978300102181
(44) Frost,D.C.;Lee,S.T.;McDowell,C.A.J.Chem.Phys.1973,59,5484.doi:10.1063/1.1679898
(45) Fock,J.H.;Gurtler,P.;Koch,E.E.Chem.Phys.1980,47,87. doi:10.1016/0301-0104(80)80023-1
(46) Ohare,P.A.G.J.Chem.Phys.1970,52,2992.doi:10.1063/ 1.1673428
(47) Breidung,J.;Thiel,W.J.Phys.Chem.A2006,110,157533.
第十八届全国电化学大会第一轮通知
由中国电化学会主办、哈尔滨工业大学承办、黑龙江大学协办的第十八届全国电化学大会将于2015年8月7-11日在黑龙江省哈尔滨市举行,本届大会主题是“支撑未来能源发展的电化学”。
全国电化学大会是国内规模最大、范围最广的电化学学术盛会和高水平的学术交流平台,每两年举办一次。本届大会围绕电化学科学和技术发展中的基础、应用和前沿问题,全面展示中国电化学领域所取得的最新研究进展和成果,深入探讨电化学领域所面临的机遇、挑战和未来发展方向,推动中国电化学学科的发展和进步,加强科研合作和技术转化,促进电化学科学与技术在能源、环境、材料等重要领域的应用。
中国电化学会热诚邀请国内外从事电化学及其相关领域的基础研究与应用开发、仪器研制以及产业界的同仁共聚美丽的哈尔滨,交流和展示最新成果。
会议预定时间:2015年8月7-11日
会议地点:哈尔滨工业大学二校区(哈尔滨市南岗区黄河路73号)
征文内容
电化学基础(包括电极过程动力学、谱学电化学、电催化等)
化学电源(包括锂离子电池、下一代储能电池、其他电池及超级电容器、燃料电池等)
环境和有机电化学(包括环境电化学、有机电合成等)
工业电化学和电化学工程(包括电解、金属腐蚀与防护、电沉积和表面处理技术等)
纳米与材料电化学
电分析化学和生物电化学(包括电化学传感器)
光电化学及新型太阳能电池(包括无机、有机光电材料与器件等)
电化学仪器与设备应用技术
联系方式
通信地址:哈尔滨工业大学1247信箱,邮编150001
联系人
投稿事宜:程新群,13895755180,chengxinqun@hit.edu.cn
学术事宜:杜春雨,0451-86403961,15846593646,duchunyu@hit.edu.cn
会务事宜:王振波,18645006176,wangzhenbo@hit.edu.cn
展览事宜:左朋建,0451-86403961,13836182469,zuopengjian@hit.edu.cn
传真:0451-86418616
本次会议论文采用在线投稿方式,关于论文格式要求及其他有关会议详情,请登陆本次大会网站http://nce.hit.edu.cn。
第十八届全国电化学大会组委会
哈尔滨工业大学化工学院
2014年11月20日
Electronic Structure and Photoionization Dissociation Process of FC(O)SNCO
ZHAO Chun-Hong1,2TONG Sheng-Rui1,*GE Mao-Fa1SUN Zheng2,*
(1Beijing National Laboratory for Molecular Sciences,State Key Laboratory for Structural Chemistry of Unstable and Stable Species,Institute of Chemistry,Chinese Academy of Sciences,Beijing 100190,P.R.China;2College of Chemistry and Materials Sciences,Hebei Normal University,Shijiazhuang 050024,P.R.China)
(Fluorocarbonyl)sulfenyl isocyanate(FC(O)SNCO)contains―C(O)S―and―NCO groups, so has an interesting electronic and molecular structure.FC(O)SNCO was prepared by the metathesis of FC(O)SCl andAgNCO.HeI photoelectron spectroscopy(PES),photoionization mass spectrometry(PIMS),and theoretical calculations were used to study the molecular and electronic structure of FC(O)SNCO.The experimental,theoretical calculations,and NBO results indicated the most stable conformer of FC(O)SNCO. In addition to the outer-valence Green's function calculation,the PES spectrum was assigned from analogous studies of similar molecules.The syn-syn non-planar conformer(δFCSN=180°)with C1symmetry was favored by the neutral molecule,and changed to the syn-syn planar structure with Cssymmetry after ionization.The experimental first vertical ionization potential mainly from the S lone pair orbital was 10.33 eV.The second to sixth ionization potentials of FC(O)SNCO were 12.44,13.23,13.77,14.78,and 15.99 eV,respectively,and were also assigned.The FC(O)+,SN+,FC(O)SN+,SNCO+,C(O)SNCO+,and FC(O)SNCO+•peaks were observed in the PIMS spectrum,the highest intensity of which was FC(O)SNCO+•.The PIMS data were analyzed basedon the PES and theoretical results.Possible ionization and dissociation processes are discussed.©Editorial office ofActa Physico-Chimica Sinica
Structure;HeI photoelectron spectroscopy;Photoionization mass spectroscopy; Theoretical calculation;Ionization and dissociation processes
O641
10.3866/PKU.WHXB201410311www.whxb.pku.edu.cn
Received:September 3,2014;Revised:October 30,2014;Published on Web:October 31,2014.
∗Corresponding authors.TONG Sheng-Rui,Email:tongsr@iccas.ac.cn;Tel:+86-10-62558682.SUN Zheng,Email:sunzheng@hebtu.edu.cn; Tel:+86-311-86268143.
The project was supported by the Strategic Priority Research Program(B)of the Chinese Academy of Sciences(XDB05010400),National Natural Science Foundation of China(41105085,21073051),and Natural Science Foundation of Hebei Province,China(B2010000368)
中国科学院战略性先导专项B(XDB05010400),国家自然科学基金(41105085,21073051)及河北省自然科学基金(B2010000368)资助项目
