NO在Ir(110)表面吸附和解离反应的泛函密度理论
2014-09-06何朝政马质璞周大伟濮春英仲志国李根全
何朝政, 马质璞, 张 帅, 周大伟, 濮春英,卢 成, 仲志国, 李 硕, 李根全
(1.南阳师范学院 物理与电子工程学院, 河南 南阳 473061;2.南阳农业职业学院 机电工程系, 河南 南阳 473000)
NO在Ir(110)表面吸附和解离反应的泛函密度理论
何朝政1, 马质璞2, 张 帅1, 周大伟1, 濮春英1,
卢 成1, 仲志国1, 李 硕1, 李根全1
(1.南阳师范学院 物理与电子工程学院, 河南 南阳 473061;
2.南阳农业职业学院 机电工程系, 河南 南阳 473000)
采用密度泛函理论(DFT)和周期性平板模型, 考察NO在Ir(110)表面上的吸附、解离及N2生成机理.计算结果表明: NO以N端向下在顶位吸附为最稳定的吸附方式, 其次是短桥位, 空位吸附最不稳定; 顶位吸附的NO在表面存在2条解离通道: 1) 直接解离通道; 2) 由初始态扩散到短桥位, 继而发生N—O键断裂生成N原子和O原子, 是NO在表面解离的主要通道; 解离后的N原子经联短桥位共吸附态发生N—N聚合反应生成N2, 在表面共存的O原子促进了N2的生成, 与实验结果相符.
密度泛函理论; NO解离; N2生成; Ir(110)表面; 过渡态
氮氧化物(NOx)是机车尾气中的主要成分之一, 对生态和生活环境及人类健康危害较大[1].目前, 通过净化器中Pt,Pd,Rh为活性金属的三效催化剂的催化作用, 可将汽车尾气中的主要污染物进行转化[2-3].但传统催化剂在催化过程中常产生N2O或NO2等污染性气体, 导致环境二次污染.由于金属Ir具有较高的催化活性和N2选择性, 因此, 关于NO在Ir表面吸附和解离的研究已引起人们广泛关注[4-7].文献[8]利用第一性原理——密度泛函理论研究了NO在Ir(100)表面的吸附位置、结构和振动频率; 文献[9-10]研究了NO在Ir(100)表面的解离及N2生成机理, 并采用微观动力学模型给出了产物的选择性信息; 文献[5]考虑了不同覆盖度下NO在Ir(111)表面的吸附; 文献[6]研究了NO在Ir(111)和Ir(211)表面的吸附和解离机理; 文献[11]通过实验研究了NO,N和O原子在金属Ir(110)表面的吸附性质, 但对于NO在Ir(110)表面的吸附结构和性质研究目前尚未见文献报道.本文采用密度泛函理论(DFT)的计算方法, 考察在0.25 ML覆盖度下NO,N和O原子在Ir(110)表面上的吸附位置及结构稳定性.
1 计算方法
本文所有电子结构计算均由VASP软件[12]完成, 计算中采用自旋极化密度泛函计算方法并结合周期性平板模型进行结构优化.选取广义梯度近似下的PW91泛函描述体系的交换相关能(GGA-PW91)[13], 采用投影扩充波(PAW)方法[14-15]描述电子-离子间的相互作用.平面波基组的动能截断值为400 eV.计算中采用的晶胞参数为0.388 nm, 该参数与实验结果0.384 nm[16]吻合较好.考虑计算的效率和精度, 采用周期性5层Ir原子的平板模型模拟金属表面, 两层平板间用1.50 nm的真空层隔开, 以确保平板间的相互作用足够小.表面采用(2×2)超晶胞, 对应0.25 ML覆盖度.表面Brillouin区k格点为3×5×1.结构优化时对下面2层Ir原子的位置固定, 吸附物和最上面3层Ir原子位置允许结构弛豫, 不考虑对称性.当作用在每个原子上的力小于每纳米0.4 eV时, 计算收敛.采用爬山构型弱助弹性带(CINEB)[17-19]方法, 搜索反应过渡态并确定最小能量路径.通过频率分析验证优化得到的稳定点构型, 即局域极小点对应全部实频, 过渡态有且只有一个虚频.能量结果均包含零点能校正(ZPE).相互作用能为共吸附结构的共吸附能与单独最稳定吸附时的吸附能之差.过渡态能量与无相互作用共吸附态的能量差用于计算双分子反应的反应能垒.
2 结果与讨论
2.1NO,N和O原子在Ir(110)表面的吸附

图1 Ir(110)表面的俯视图Fig.1 Top view of Ir(110) surface
2.1.1 NO吸附 分别考虑NO在顶位、长桥位、短桥位和空位的4种吸附模式, 如图1所示.其中T,H,SB,LB分别表示顶位、空位、短桥位和长桥位, 黑色和白色的金属球分别表示第一层和第二层原子.计算不同模型的吸附能, NO分子在Ir(110)表面上的吸附位置、构型参数、振动频率及吸附能列于表1.吸附能的表达式为
其中:Eads为吸附能;Eadsorbate-surface,Esurface和Egas分别表示吸附后体系的总能量、干净弛豫底物的能量和吸附前气相分子的能量.
由表1可见: 顶位、长桥位和短桥位吸附的NO分子, 其N—O键长从气相时的0.116 9 nm分别拉长至0.118 1,0.121 1,0.121 2 nm, N原子与Ir原子的距离分别为0.178 5,0.212 0,0.197 7 nm, 相应的吸附能分别为-2.85,-2.00,-2.58 eV, 零点能校正后分别为-2.78,-1.94,-2.52 eV; 在0.25 ML覆盖度下, NO分子在Ir(110)表面上最稳定的吸附位是顶位, 其次是短桥位, 长桥位的稳定性较小, 未发现空位吸附结构; N—O键在3种吸附位置的伸缩振动频率分别为1 828,1 531,1 574 cm-1; 在短桥位比顶位吸附NO分子的键长略长, 即短桥位吸附NO的N—O键相对较弱, 比顶位吸附更易发生解离.
2.1.2 N原子吸附 N原子在Ir(110)表面吸附的结果表明: N原子可稳定吸附在Ir(110)表面的短桥位、空位、长桥位和顶位上, 吸附能分别为-5.61,-5.10,-4.69,-4.63 eV, 零点能校正后的吸附能分别为-5.52,-5.02,-4.62,-4.55 eV; 最稳定吸附位是短桥位; 比次稳定的空位结构能量低0.51(0.50)eV, 即N原子易于吸附在短桥位上; 在最稳定的短桥位结构中, N—Ir键长为0.186 2 nm, N原子离表面的垂直距离为0.125 8 nm.

表1 DFT计算的结构参数、零点能校正后的吸附能及NO分子在Ir(110)表面上不同吸附位置的频率*Table 1 DFT calculated structural parameters, ZPE corrected adsorption energies (Eads) and predicted υ(NO) for NO adsorption on Ir(110) surface
2.1.3 O原子吸附 O原子在Ir(110)表面吸附的结果表明: O原子可稳定吸附在Ir(110)表面的短桥位、顶位、空位和长桥位上, 吸附能分别为-5.73,-5.05,-5.01,-4.79 eV, 零点能校正后的吸附能分别为-5.66,-4.99,-4.95,-4.73 eV; 最稳定吸附位是短桥位, 比次稳定的顶位结构能量低0.68(0.67)eV, 即O原子易于吸附在短桥位上; 在最稳定的短桥位结构中, O—Ir键长为0.193 1 nm, O原子与表面的垂直距离为0.136 3 nm.
2.2NO在Ir(110)表面上的解离路径
以顶位吸附的NO(结构1)为初始反应物可得到2种不同的解离路径, 2个反应路径中的过渡态、末态结构和相应的反应势能面如图2所示.
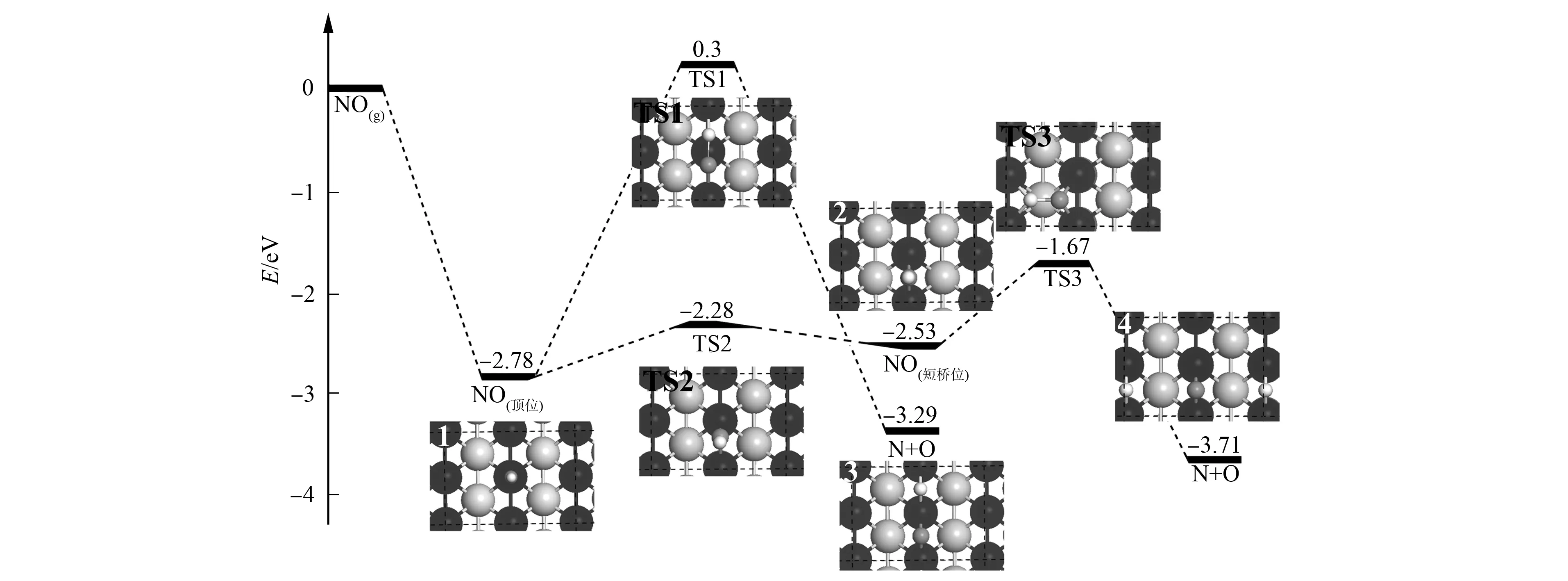
图2 NO在Ir(110)表面上的解离势能面Fig.2 Potential energy diagram for NO dissociation on Ir(110)
由图2可见: 吸附在桥位的NO分子经过渡态TS1发生直接解离, 末态产物(结构3)的N和O原子分别吸附在相邻短桥位上, 与NO初始吸附的顶位Ir原子距离分别为0.191 1,0.197 9 nm; 在过渡态TS1中, N—O键长由初始结构的0.118 1 nm伸长至0.176 5 nm, N和O原子分别移至相邻的短桥位上, 与最近邻的顶位Ir原子距离分别为0.182 5,0.200 6 nm; 过渡态的虚频值为660.1 cm-1, 对应N—O的伸缩振动, 该过程需克服的反应能垒为3.18 eV, 反应放热0.56 eV; 零点能校正后的能垒和反应热分别为3.08,0.51 eV; 顶位吸附的NO分子先经过渡态TS2由顶位扩散至短桥位, 形成短桥位吸附的结构2, 该过程扩散能垒为0.53 eV; 零点能校正后能垒为0.50 eV; 在TS2中, N—O键长由顶位时的0.118 1 nm伸长至0.120 0 nm, N—Ir键长为0.184 5 nm, 过渡态的虚频为322.8 cm-1; 短桥位结构中的N—O键长为0.121 2 nm, 即N—O键强度明显减弱, 因此, 短桥位吸附的NO易于越过低垒过渡态TS3发生解离, 能垒仅为0.92 eV, 形成产物为具有联桥位的N和O原子的共吸附结构4, 整个过程体系放热1.17 eV; 零点能校正后的能垒和反应热分别为0.86,-1.18 eV; 过渡态TS3中的N—O键长为0.158 8 nm, 基本完全断开; 过渡态虚频为464.8 cm-1, 对应N—O伸缩振动.
2.3N2生成反应

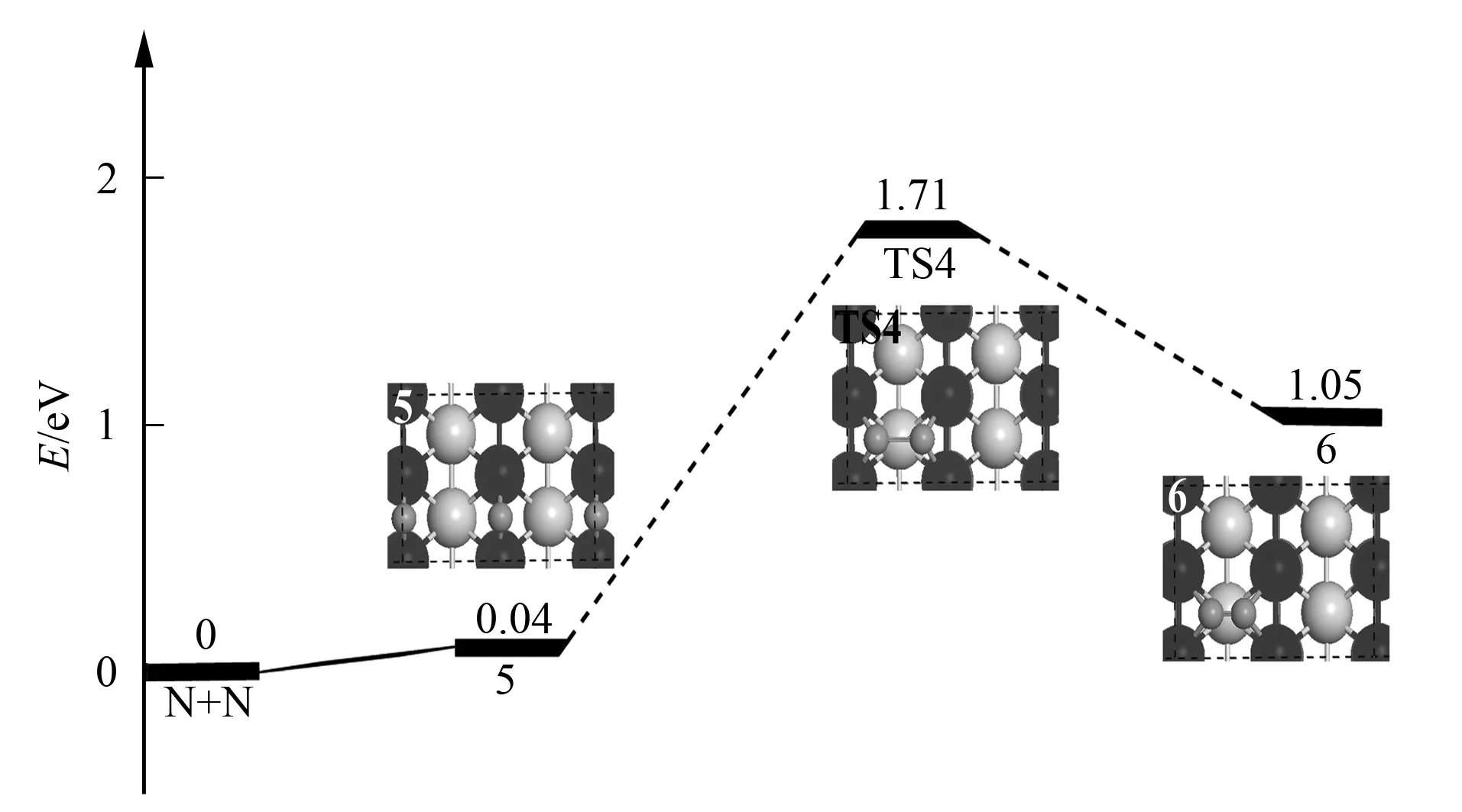
图3 Ir(110)表面上N原子聚合反应的势能面Fig.3 Potential energy diagram for the N adatoms combination reaction on Ir(110)

图4 O预吸附Ir(110)表面上N原子聚合反应的势能面Fig.4 Potential energy diagram for the N adatoms combination reaction on O-predosed Ir(110)
综上, 本文利用第一性原理考察了NO分子在Ir(110)表面的吸附、解离和N2的生成过程, 可得如下结论: 1) 在Ir(110)表面, NO以N端向下在顶位吸附为最稳定的吸附方式, 吸附能为-2.78 eV, 其次是短桥位, 吸附能为-2.52 eV, 空位吸附最不稳定; 2) NO在Ir(110)表面存在2条解离通道: 主要解离通道为顶位吸附的NO分子先克服0.53 eV的扩散能垒至较易解离的短桥位吸附结构, 再需0.86 eV的能量即可解离生成N与O原子联桥位共吸附结构; 直接解离的反应能垒较高, 为3.08 eV; 3) 在Ir(110)表面存在O原子可促进N—N聚合反应发生, 与实验结果一致.
[1]Brown W A, King D A.NO Chemisorption and Reactions on Metal Surfaces: A New Perspective [J].J Phys Chem B, 2000, 104: 2578-2595.
[2]Inoue T, Tomishige K, Iwasawa Y.Characterization of Pt-Sn/SiO2Catalysts and the Role of Sn in NO-Hydrocarbon Reactions [J].J Chem Soc Faraday Trans, 1996, 92: 461-467.
[3]Burch R, Millington P J, Walker A P.Mechanism of the Selective Reduction of Nitrogen Monoxide on Platinum-Based Catalysts in the Presence of Excess Oxygen [J].Appl Catal B: Environ, 1994, 4(1): 65-94.
[4]Fujitani T, Nakamur I, Takahashi A, et al.Kinetics and Mechanism of NO Reduction with CO on Ir Surfaces [J].J Catal, 2008, 253: 139-147.
[5]ZENG Zhenhua, Juarez L F, Silvab D, et al.Theory of Nitride Oxide Adsorption on Transition Metal (111) Surfaces: A First-Principles Investigation [J].Phys Chem Chem Phys, 2010, 12(10): 2459-2470.
[6]Liu Z P, Jenkins S J, King D A.Step-Enhanced Selectivity of NO Reduction on Platinum-Group Metals [J].J Am Chem Soc, 2003, 125(48): 14660-14661.
[7]Krekelberg W P, Greeley J, Mavrikakis M.Atomic and Molecular Adsorption on Ir(111) [J].J Phys Chem B, 2004, 108(3): 987-994.
[8]Khatua S, Liu Z P, King D A.NO Restructuring of Surface Ir and Bond Formation to Preadsorbed O on Ir(100) at 95 K [J].Surf Sci, 2005, 584: 214-224.
[9]He C Z, Wang H, Zhu P, et al.Adsorption and Dissociation of NO on Ir(100): A First-Principles Study [J].J Chem Phys, 2011, 135(20): 204707.
[10]何朝政, 王会, 淮丽媛, 等.NO在氧预吸附Ir(100)表面吸附和解离的第一性原理研究 [J].高等学校化学学报, 2013, 34(4): 946-951.(HE Chaozheng, WANG Hui, HUAI Liyuan, et al.First-Principles Study on NO Adsorption and Dissociation on O-Predosed Ir(100) [J].Chem J Chinese University, 2013, 34(4): 946-951.)
[11]Wolf C A, de, Bakker J W, Wouda P T, et al.Evidence of Repulsive Interactions between NO, O, and N on Ir(110).A Fast X-Ray Photoelectron Spectroscopy Study [J].J Chem Phys, 2000, 113(23): 10717-10722.
[12]Kresse G, Hafner J.AbinitioMolecular Dynamics for Liquid Metals [J].Phys Rev B, 1993, 47(1): 558-561.
[13]Perdew J P, Chevary J A, Vosko S H, et al.Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation [J].Phys Rev B, 1992, 46(11): 6671-6687.
[14]Blöchi P E.Projector Augmented-Wave Method [J].Phys Rev B, 1994, 50(24): 17953-17979.
[15]Kresse G, Joubert D.From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method [J].Phys Rev B, 1999, 59(3): 1758-1775.
[16]Kittel C.Introduction to Solid State Physics [M].New York: Wiley, 1996.
[17]Henkelman G, Uberuaga B P, Jónsson H.A Climbing Image Nudged Elastic Band Mehtod for Finding Saddle Points and Minimum Energy Paths [J].J Chem Phys, 2000, 113(22): 9901-9904.

[19]王兴, 濮春英, 靳希联.压力下超导LiBe合金热动力学性质的第一原理计算 [J].吉林大学学报: 理学版, 2014, 52(4): 816-819.(WANG Xing, PU Chunying, JIN Xilian.First-Principle Calculations of Thermal Dynamical Properties of Superconducting LiBe Alloy under Pressure [J].Journal of Jilin University: Science Edition, 2014, 52(4): 816-819.)
DensityFunctionalTheoryofNOAdsorptionandDissociationonIr(110)Surface
HE Chaozheng1, MA Zhipu2, ZHANG Shuai1, ZHOU Dawei1, PU Chunying1,
LU Cheng1, ZHONG Zhiguo1, LI Shuo1, LI Genquan1
(1.CollegeofPhysicsandElectronicEngineering,NanyangNormalUniversity,Nanyang473061,HenanProvince,China; 2.DepartmentofMechanicalandElectricalEngineering,NanyangVocationalCollegeofAgriculture,Nanyang473000,HenanProvince,China)
Density functional theory and periodic slab model were adopted to systemically study NO adsorption, dissociation and N2formation on Ir(110) surface.The results show that the most of stable sites are the top sites with the nitrogen bonding to Ir atoms, the short-bridge sites are less stable and the hollow sites are the least of all.There are two paths for the dissociation of NO adsorbed on the top sites: One is direct dissociation; and the other is diffusion from top site to short-bridge site firstly and then dissociation.The results of potential energy surface show that the second path is more favorable than the first one, and is the primary path for NO dissociation.Two N atoms from the dissociated NO form a di-short-bridge structure to recombine into N2.In addition, the pre-dosed O atom enhances the N atoms combination reaction, which is consistent well with experiment result.
density functional theory; NO dissociation; N2formation; Ir(110) surface; transition state
2014-05-19.
何朝政(1982—), 男, 汉族, 博士, 讲师, 从事表面催化的研究, E-mail: hecz2008@gmail.com.通信作者: 马质璞(1962—), 女, 汉族, 讲师, 从事表面催化的研究, E-mail: Mzp620315@163.com; 李根全(1961—), 男, 蒙古族, 博士, 教授, 从事理论物理和凝聚态物理的研究, E-mail: 13783775918@163.com.
国家自然科学基金(批准号: U1404216; U1404608; U1304612; 11247222)、河南省科技厅科技攻关项目(批准号: 142102210478; 132102210481)、河南省中青年骨干教师资助项目(批准号: 2012GGJS-152)和南阳师范学院科研项目(批准号: ZX2014088; ZX2012018; ZX20100011).
O521.2
A
1671-5489(2014)06-1301-05
10.13413/j.cnki.jdxblxb.2014.06.37
王 健)
