心肌病的遗传学研究进展
2014-07-16刘雯综述刘文玲审校
刘雯 综述 刘文玲 审校
(北京大学人民医院心脏中心,北京100044)
心肌病是非冠心病、高血压、瓣膜病和先天性心脏病等原因所引起的心肌结构及功能异常,可表现为心脏机械活动或电活动的异常,包括遗传性和非遗传性心肌病。遗传性心肌病包括原发于心肌的扩张型心肌病(DCM)、肥厚型心肌病(HCM)、限制型心肌病(RCM)、致心律失常型右室心肌病(ARVC)、左室致密化不全(LVNC)和累及心肌的系统性疾病如Fabry病和Kearns-Sayre综合征[1]。近年来,在心肌病遗传学研究方面取得了很大进展,目前已发现了相关的多个致病基因和突变,并在发病机制方面进行了探索。但基因突变和疾病的发生和预后之间的关系仍是尚未解决的难题。临床基因筛查,作为遗传性疾病诊断的一个重要手段,在协助确诊心肌病,发现亚临床病人和筛查先证者的家属方面有重要作用,但仍面临着很多问题。现对遗传性心肌病的临床特点、发病机制、遗传学和基因筛查的研究进展进行综述。
1 肥厚型心肌病
肥厚型心肌病是以不对称性左心室肥厚为特征的常见的遗传性心肌病,以常染色体显性遗传为主,是青年人猝死的最常见原因。其在人群中的发病率约为1∶500,男性多于女性。组织学表现为心肌细胞肥大,排列紊乱伴间质纤维灶的形成。临床表现具有明显的异质性,从无症状、胸闷、心悸到晕厥。肥厚型心肌病可在婴儿到老人的任何一个阶段发病,大部分患者预后良好,可达到正常预期寿命,部分患者后期出现心房颤动栓塞、恶性室性心律失常、心力衰竭等不良事件[2]。
1.1 遗传学特征
1990年报道了肥厚型心肌病的第一个致病基因MYH7[3]。目前认为肥厚型心肌病是种肌小节病。最主要的致病基因主要编码心脏肌球蛋白重链(MYH7)和心脏肌球蛋白结合蛋白C(MYBPC3),分别占家族性肥厚型心肌病患者的30% ~50%和20%[4]。其他的致病基因包括主要编码粗肌丝蛋白[肌球蛋白重链MYH6、肌球蛋白轻链(MYL2、MYL3)],细肌丝蛋白[肌动蛋白 ACTC、原肌球蛋白 TPM1、心脏肌钙蛋TNNT2、心脏肌钙蛋白 TNNⅠ3、肌钙蛋白 C(TNNC1)],装配蛋白基因(肌联蛋白TTN)。其中,TNNT2、TNNⅠ3、TPM1 突变占患者的 1% ~5%[5-8]。新报道的致病基因Z蛋白相关基因和钙离子相关蛋白,在人群中的突变频率低,主要发生于散发人群,与肥厚型心肌病发病的关系不明确。在基因型阳性的患者中,3%~5%的患者同时携带两个突变或多个突变[6-7](表1)。不同种族人群中基因突变的频率有明显的差异,波动在24% ~63%(瑞典人群24%,欧洲人群63%[5,8]。常见的突变热点包括:MYH7 基因上Arg403、Arg453、Arg663、Gly741、Arg719 和 Asp778。MYBPC基因上 Arg502和 IVS20-2,TNNT2基因上Arg92和 Arg58。
肥厚型心肌病基因型和表型的关系至今仍未完全阐明,基因型某种程度上会影响疾病的严重程度和预后。Olivotto等[9]研究发现在基因型阳性的患者出现左室收缩和舒张功能障碍的危险性大。基因型阳性的患者发生心脏性死亡、致命性脑卒中和心力衰竭的危险性明显增加。且多个突变的携带者较单个突变携带者预后差[6]。
既往认为MYBPC3突变携带者发病晚,心肌肥厚程度轻,MYH7突变的携带者发病早,症状重,心肌肥厚程度重。TNNT2突变的携带者往往临床症状较轻,但猝死率高,尤其是在心肌肥厚程度较轻的青少年中[10-11]。长期的随访研究发现 MYBPC3、MYH7 和TNNT2的突变携带者的预后无统计学差异[6]。早期所定义的恶性突变包括 MYH7-R403Q、TNNT2-I79N等在不同个体有不同的临床表现[10,12]。这些研究说明不能通过特定的基因或突变来判断肥厚型心肌病的预后。
基因突变引起心肌肥厚的机制未完全阐明。目前认为基因突变导致氨基酸的改变,形成“毒肽”渗入到肌小节中干扰正常的肌小节的组装和收缩功能,导致肌纤维的排列紊乱[13]。部分突变,如移码突变和无义突变,常见于MYBPC3基因,形成截断蛋白或不稳定的蛋白,不能参与到肌小节的组装中,导致单倍体不足[14];动物实验提示在突变的模型中Ca的敏感性和肌动蛋白激活的三磷酸腺苷(ATP)酶的活性有所改变,但不同实验报道有所差异[13,15]。亦有研究发现突变的MYBPC3会影响下游的几个与心肌的代谢通路相关基因的编码。另外,突变的MYBPC3可能会影响泛素化导致泛素对其他的蛋白底物的清除出现障碍,最终导致心肌肥厚。
1.2 基因筛查
肥厚型心肌病的基因筛查主要针对MYBPC3、MYH7、TNNⅠ3、TNNT2和TPM1这5个主要的致病基因。目前,指南推荐对临床表现不典型病例或者疑似其他遗传性疾病的患者进行筛查,以协助诊断。对于携带有基因阳性的先证者的家属亦推荐进行相关基因筛查,明确家族中有潜在的患者,以进行进一步的随访和干预[2]。
2 致心律失常型右室心肌病
致心律失常型右室心肌病,是一种主要累及右心室的遗传性心肌病。1977年由Fontaine等首先报道,其病理特征为进行性右心室心肌坏死和脂肪纤维组织增生并伴随炎症反应,可同时累及双室。致心律失常型右室心肌病的典型临床常表现为心律失常,随着疾病的进展会出现形态学改变甚至出现心力衰竭。致心律失常型右室心肌病心律失常以左束支阻滞的室性心动过速为特征,是导致晕厥和猝死的直接原因。
致心律失常型右室心肌病在人群中的患病率为0.5% ~1.0%,其发病呈现一定的家族性,其中50%~70%的病例是家族性的[16]。致心律失常型右室心肌病主要为常染色体显性遗传,根据其致病基因的不同分为相关亚型,其中Naxos综合征为常染色体隐性遗传,其外显率不一。
2.1 遗传学特征
目前已发现了8个致心律失常型右室心肌病的致病基因,其中5个基因均编码桥粒蛋白 (表1)。基因突变造成的桥粒蛋白功能不全可能是该疾病的“最后的共同通路”。其中,PKP2是致心律失常型右室心肌病最常见的致病基因,占致心律失常型右室心肌病突变的10% ~52%。其次为 DSP、DSG2和 DSC2,其突变频率差异大,与种族、地域等有密切关系。JUP基因上的突变相对较少[17]。另外,有研究报道致心律失常型右室心肌病的发病和非桥粒蛋白相关,如跨膜蛋白43(TMEM43)、TGFb-3 和 RyR2 基因[18-20]。近年来,亦有研究报道致心律失常型右室心肌病的发病与连接素蛋白TTN基因相关[21]。
目前,致心律失常型右室心肌病的发病机制并未完全阐明,一些假说认为基因突变会导致细胞连接受损或者干扰wnt/β-catenin信号通路等[22]。但这些学说并不能完全解释桥粒蛋白突变和心脏的电生理改变之间的关系。近年来发现,致心律失常型右室心肌病的患者中,Nav1.5、连接蛋白43和斑珠蛋白几个蛋白表达减少,提示斑珠蛋白和其他的桥粒蛋白可能参与了细胞骨架依赖的离子通道的转运,影响到连接蛋白43的定位和 Nav1.5通道。包括连接蛋白43、PKP2、斑珠蛋白和其他支架蛋白在内的闰盘蛋白重构可能会导致钠通道的减少,从而引起心律失常[23]。
2.2 基因筛查
致心律失常型右室心肌病患者中约有50%的患者携带桥粒蛋白相关突变,由于基因突变致病的机制尚未明确,基因筛查在致心律失常型右室心肌病患者中主要应用于协助疑诊的患者确诊,或对基因型阳性的先证者的家族成员进行筛查。
3 扩张型心肌病
扩张型心肌病是临床上一类既有遗传因素又有非遗传因素造成的复合型心肌病,以单侧或双侧心腔扩大和心室收缩功能障碍为主要特征。发病率为1∶2 500,大部分为散发病人,只有35%的患者有家族史。其组织学特点包括心肌坏死和纤维化。临床上,个体表现差异性大,表现为不同程度的心脏扩大和心功能障碍,部分患者伴有心脏传导疾病。部分患者可表现为心律失常,猝死率高。
3.1 遗传学特征
扩张型心肌病的遗传方式多种多样,包括常染色体显性遗传、染色体隐性遗传、X连锁的和线粒体遗传等方式,主要以常染色体显性遗传为主。其致病基因多,研究发现至少15个基因与扩张型心肌病的发病相关,这些基因主要编码肌小节相关蛋白和细胞骨架蛋白(表1)[24-25]。其中,TTN基因突变在家族性和散发病人的突变频率分别是25%和18%,被认为是扩张型心肌病最常见的突变基因[26]。扩张型心肌病的某些特定的表型和特定的基因突变相关联,如核纤层蛋白A/C(LMNA)和SCN5A上的突变和心脏传导系统疾病相关,X连锁遗传Dystrophin和Taffazin基因的突变和肌肉疾病相关。目前基因突变致病的机制还不明确,研究认为突变可能影响ATP的利用率、Ca2+敏感性以及肌动和肌球蛋白的相互作用,从而导致心肌收缩力减弱[24,27]。除了遗传因素外,其他因素如持续性病毒感染和自身免疫反应也参与了扩张型心肌病的发病。
3.2 基因筛查
与肥厚型心肌病不同,扩张型心肌病致病基因多,突变频率低,基因筛查的效率低,花费高,家族性扩张型心肌病并不推荐常规做基因筛查。但对于有些特殊的表型,如扩张型心肌病伴有心脏传导系统障碍或肌肉疾病,可对特定的基因(LMNA、Dystrophin、Taffazin)进行筛查。
4 限制型心肌病
限制型心肌病是以心室心内膜纤维化、心室舒张功能障碍为特征的一类心肌病,其心室大小、室壁厚度和心室收缩功能大致正常。临床相对少见。病理学表现为心内膜心肌纤维化,心肌细胞溶解、变性。限制型心肌病往往起病隐匿,主要表现为静脉回流障碍和心排血量减少的症状和体征[1]。
遗传方式以常染色体显性遗传为主,也可表现为常染色体隐性遗传。限制型心肌病的发病主要与编码结蛋白(desmin)的基因DES突变相关。部分病例报道在限制型心肌病患者中发现编码肌小节的基因TNNⅠ3、TNNT2、MYH7和ACTC1突变。基因筛查在限制型心肌病的应用较少,主要对基因型阳性的先证者的家属进行筛查[28-29]。
5 心肌致密化不全
心肌致密化不全是一种遗传相关的、心室内肌小梁异常粗大、深隐窝交错的先天性心脏畸形。该病患者的主要临床表现有心功能不全、心律失常和栓塞。心电图上可表现为多种心律失常,这可能与肌小梁及分支的连接不规则导致的心肌电生理不稳定有关系。此外,由于肌小梁间隐窝的存在,血液在此流速减慢、瘀滞,极容易形成血栓,进而引发脑栓塞、肺栓塞、肠系膜血管栓塞等问题[30]。
心肌致密化不全发病呈现散发性和家族性两大类,其中家族发病者占18% ~25%[31]。现已发现,心肌致密化不全的遗传方式可有常染色体显性遗传、X连锁遗传、线粒体遗传等。目前已发现有TAZ、LDB3、LMNA、DTNA等多个基因可能和心肌致密化不全相关(表1)[32-33]。值得注意的是,这些基因突变还与多种心肌病、骨骼肌肉病、线粒体疾病相关。如此复杂的遗传学背景,也解释了很多心肌致密化不全往往合并多种遗传性疾病,其确切的遗传学机制仍待进一步研究。
6 Fabry病
Fabry病又称为弥漫性体血管角质瘤,是一种家族遗传性的磷脂累积病,该病于1984年由 Johann Fabry和William Anderson首次报道。Fabry病是一种性染色体隐性遗传病,致病基因GLA位于X染色体长臂Xq22。相关基因突变导致 α-半乳糖苷酶 A(α-GalA)部分或者完全缺失,从而使得其代谢底物神经酰胺三聚己糖(Gb3)蓄积于神经系统、肾脏以及心血管系统,造成相关系统的病变。心脏损害包括心肌损害、心脏瓣膜损害和心律失常[34]。本病发病率低,常在青少年期发病,主要出现在男性儿童,不同地区间发病率有很大的差异。
Fabry可分为经典型和迟发型两类。经典型多为男性半合子,此时α-GalA活性明显下降甚至完全缺失,从而出现肾脏、心血管等多系统的累及,根据病情进展程度,又可将本病分为三个时期:(1)第一期出现在儿童期,表现为四肢烧灼样疼痛、皮温升高、皮下多发血管瘤、角膜混浊等;(2)第二期主要表现为肾脏损坏,出现蛋白尿、血尿、肾功能下降等;(3)第三期发生严重肾损害,并累及心脑血管系统,可因肾衰竭或严重的心脑血管并发症死亡。迟发型患者α-GalA活性部分下降,临床症状较轻,女性杂合子多为该型。
7 Kearns-Sayre综合征
Kearns-Sayre综合征,又称为眼肌麻痹综合征、眼肌麻痹-色素性视网膜炎-心脏传导阻滞综合征、眼咽肌营养不良等,是线粒体脑肌病中唯一累及心脏传导系统的疾病。57%的Kearns Sayre综合征患者会有心血管系统的损害,表现为晕厥、充血性心力衰竭、心脏停搏等。本病可累及房室结、希氏束远端等部位,患者多在眼部病变之后出现症状轻微的传导阻滞,并进行性加重,逐渐发展为第二度甚至第三度房室传导阻滞,严重者可能有猝死的风险。此外,还有患者可因QT间期延长、三分支阻滞而诱发尖端扭转型室性心动过速,这也是一大致死原因。
目前认为,Kearns-Sayre综合征的发病机制主要与线粒体基因的片状缺失及点突变相关,最常见的是线粒体DNA 8482~13459位之间4979 bp片段的缺失。线粒体DNA突变使得氧化代谢过程中所必需的酶或载体代谢障碍,糖原和脂肪酸不能进入线粒体或不能被充分利用,ATP合成障碍进一步导致能力供应不足,最终影响多系统的功能活动[35]。在该病的发病过程中,除了线粒体DNA参与外,核基因组也起着非常重要的作用,但具体机制尚不明确,这也是今后的研究方向和热点。由于其特殊的发病机制,因此本病具有明显的母系遗传的特点。
8 基因筛查在遗传性心肌病中的应用
遗传性心肌病的基因筛查目前已在很多大型的商业实验室开展。肥厚型心肌病作为最常见的遗传性心肌病,筛查的阳性率较高,在家族性心肌病的患者为60%,散发性心肌病的患者为40%,基因筛查在该疾病应用最为广泛。
在诊断方面,对临床表现不典型病例,基因型阳性可协助疾病的确诊,但由于只有部分患者携带相关突变,基因型阴性亦不能排除诊断。对于模棱两可的病例,基因筛查还可协助排除其他诊断。如代谢性心肌病可表现为不明原因的左室肥厚,其临床表现与肥厚型心肌病难于区分,但这些患者,编码蛋白激酶的γ2的调节亚基的基因PRKAG2和编码X连锁的与膜蛋白相关的溶酶体基因LAM有突变。对于基因型阳性的先证者,对其家属进行相关基因的筛查可发现有患病危险的个体。遗传性心肌病在临床表现上往往相互重叠,如晚期的肥厚型心肌病和致心律失常型右室心肌病在形态学上可表现为心室腔的扩大。早期肥厚型心肌病在有明显的心肌肥厚之前可表现为单纯的舒张功能障碍,与限制型心肌病相似;同时,其致病基因亦相互重叠,例如,肌小节基因突变最开始认为只与肥厚型心肌病的发病相关,但研究发现肌小节基因突变也与扩张型心肌病、限制型心肌病和左室致密化不全的发病相关,Z盘基因与扩张型心肌病和肥厚型心肌病发病均相关。TTN被认为是扩张型心肌病最常见的突变基因,目前发现与致心律失常型右室心肌病的发病亦相关。由于其致病基因缺乏特异性,在临床表现不典型时,基因筛查较难提供特异性的疾病诊断。
基因筛查可对疾病的临床预后提供一定指导。总体而言,基因型阳性的患者其预后较基因型阴性的患者预后差,一些特定的突变与特定的表型相关,如扩张型心肌病中,LMNA和SCN5A和心脏传导系统疾病相关,X连锁遗传Dystrophin和Taffazin与肌肉疾病相关。但由于遗传性心肌病是一组多基因疾病,具有遗传异质性,多种因素会影响疾病的预后,包括环境、修饰基因、感染和免疫等。疾病的预后不能单独取决于某个基因上的特定的基因或特定突变。即使在同一家系中携带同一突变的个体也可能出现不同的临床表现和预后。因此,如何通过基因型对个体的临床预后进行预测是基因筛查应用于临床的一大困境。
人群中往往亦存在基因的变异,这些变异并不致病,但正常变异的存在会对基因筛查的结果产生干扰,当在有临床表型的个体中发现基因突变时,很难确定该突变是致病的或为正常变异。随着遗传学研究的深入,已有大量的遗传性心肌病相关的基因和突变被报道,很多突变仅在少数散发先证者中被发现,往往缺乏严格的遗传支持,我们很难确定该突变的致病性。这部分介于良性突变和致病性突变之间的突变被定义为不清楚临床意义的变异(VUS)。如何确实地区分VUS和致病性突变,提供准确的遗传学建议是基因筛查中的另一大难题,对于基因型阳性者的治疗选择亦有重要的指导意义。
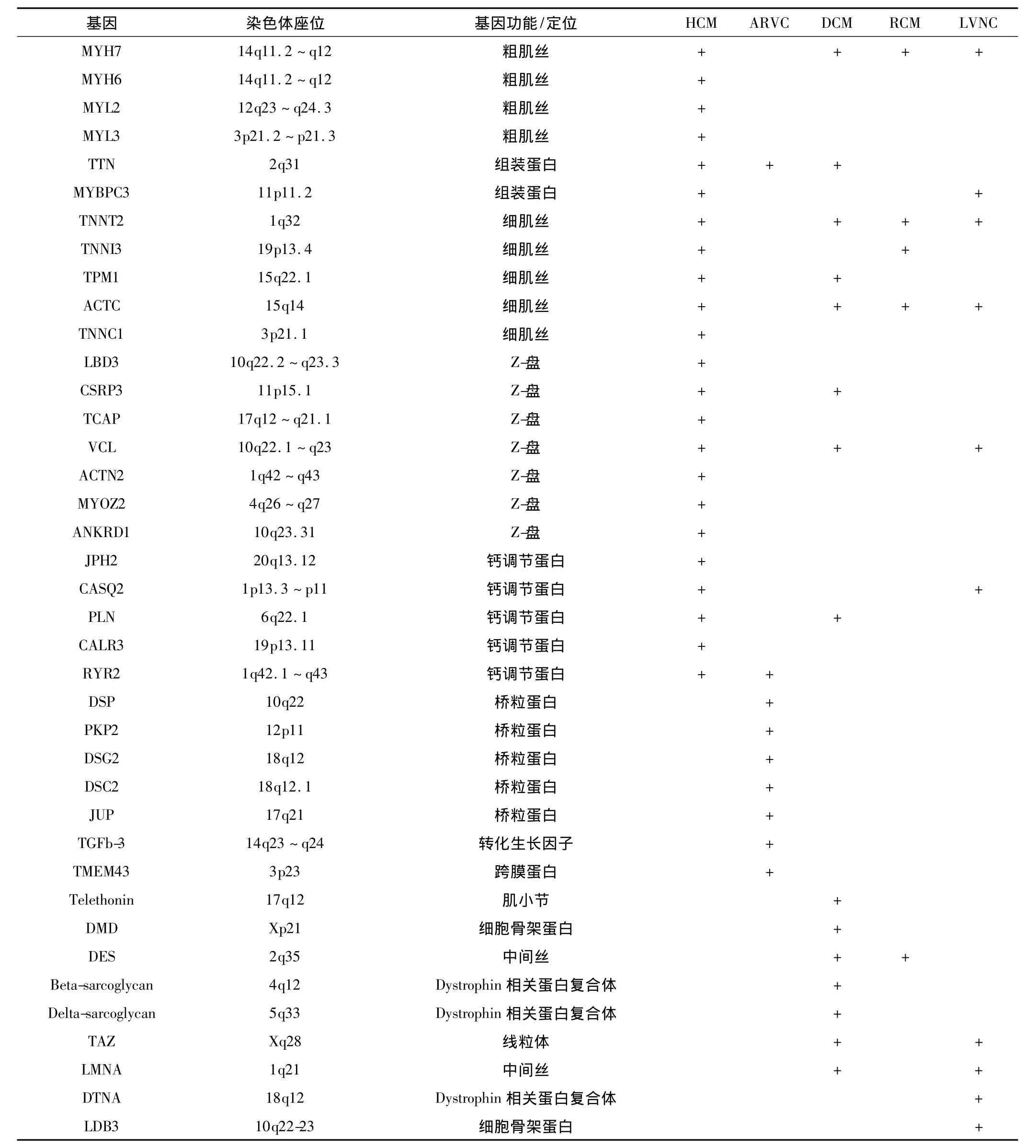
表1 遗传性心肌病相关基因
[1]Elliott P,Andersson B,Arbustini E,et al.Classification of the cardiomyopathies:a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases[J].Eur Heart J,2008,29(2):270-276.
[2]Gersh BJ,Maron BJ,Bonow RO,et al.2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy:a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines[J].J Thorac Cardiovasc Surg,2011,142(6):e153-203.
[3]Geisterfer-Lowrance AA,Kass S,Tanigawa G,et al.A molecular basis for familial hypertrophic cardiomyopathy:a beta cardiac myosin heavy chain gene missense mutation[J].Cell,1990,62(5):999-1006.
[4]Marian AJ,Roberts R.The molecular genetic basis for hypertrophic cardiomyopathy[J].J Mol Cell Cardiol,2001,33(4):655-670.
[5]Richard P,Charron P,Carrier L,et al.Hypertrophic cardiomyopathy:distri-bution of disease genes,spectrum of mutations,and implications for a molecular diagnosis strategy[J].Circulation,2003,107(17):2227-2232.
[6]van Driest SL,Vasile VC,Ommen SR,et al.Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy[J].J Am Coll Cardiol,2004,44(9):1903-1910.
[7]van Driest SL,Jaeger MA,Ommen SR,et al.Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy[J].J Am Coll Cardiol,2004,44(3):602-610.
[8]Morner S,Richard P,Kazzam E,et al.Identification of the genotypes causing hypertrophic cardiomyopathy in northern Sweden[J].J Mol Cell Cardiol,2003,35(7):841-849.
[9]Olivotto I,Girolami F,Ackerman MJ,et al.Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy[J].Mayo Clin Proc,2008,83(6):630-638.
[10]Watkins H,Rosenzweig A,Hwang DS,et al.Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy[J].N Engl J Med,1992,326(17):1108-1114.
[11]Song L,Zou Y,Wang J,et al.Mutations profile in Chinese patients with hypertrophic cardiomyopathy[J].Clin Chim Acta,2005,351(1-2):209-216.
[12]Seggewiss H,Gleichmann U,Faber L.The management of hypertrophic cardiomyopathy[J].N Engl J Med,1997,337(5):349-350.
[13]Olsson MC,Palmer BM,Stauffer BL,et al.Morphological and functional alterations in ventricular myocytes from male transgenic mice with hypertrophic cardiomyopathy[J].Circ Res,2004,94(2):201-207.
[14]Rottbauer W,Gautel M,Zehelein J,et al.Novel splice donor site mutation in the cardiac myosin-binding protein-C gene in familial hypertrophic cardiomyopathy.Characterization Of cardiac transcript and protein[J].J Clin Invest,1997,100(2):475-482.
[15]Lankford EB,Epstein ND,Fananapazir L,et al.Abnormal contractile properties of muscle fibers expressing beta-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy[J].J Clin Invest,1995,95(3):1409-1414.
[16]Ackerman MJ,Priori SG,Willems S,et al.HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies:this document was developed as a partnership between the Heart Rhythm Society(HRS)and the European Heart Rhythm Association(EHRA)[J].Europace,2011,13(8):1077-1109.
[17]Jacob KA,Noorman M,Cox MG,et al.Geographical distribution of plakophilin-2 mutation prevalence in patients with arrhythmogenic cardiomyopathy[J].Neth Heart J,2012,20(5):234-239.
[18]Merner ND,Hodgkinson KA,Haywood AF,et al.Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant,lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene[J].Am J Hum Genet,2008,82(4):809-821.
[19]Beffagna G,Occhi G,Nava A,et al.Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1[J].Cardiovasc Res,2005,65(2):366-373.
[20]Tiso N,Stephan DA,Nava A,et al.Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2(ARVD2)[J].Hum Mol Genet,2001,10(3):189-194.
[21]Taylor M,Graw S,Sinagra G,et al.Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes[J].Circulation,2011,124(8):876-885.
[22]Garcia-Gras E,Lombardi R,Giocondo MJ,et al.Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy[J].J Clin Invest,2006,116(7):2012-2021.
[23]Shaw RM.Reduced sodium channels in human ARVC[J].Heart Rhythm,2013,10(3):420-421.
[24]Kamisago M,Sharma SD,DePalma SR,et al.Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy[J].N Engl J Med,2000,343(23):1688-1696.
[25]Villard E,Duboscq-Bidot L,Charron P,et al.Mutation screening in dilated cardiomyopathy:prominent role of the beta myosin heavy chain gene[J].Eur Heart J,2005,26(8):794-803.
[26]Herman DS,Lam L,Taylor MR,et al.Truncations of titin causing dilated cardiomyopathy[J].N Engl J Med,2012,366(7):619-628.
[27]Morimoto S,Lu QW,Harada K,et al.Ca(2+)-desensitizing effect of a deletion mutation Delta K210 in cardiac troponin T that causes familial dilated cardiomyopathy[J].Proc Natl Acad Sci U S A,2002,99(2):913-918.
[28]Kaski JP,Syrris P,Burch M,et al.Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes[J].Heart,2008,94(11):1478-1484.
[29]Mogensen J,Kubo T,Duque M,et al.Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations[J].J Clin Invest,2003,111(2):209-216.
[30]Pignatelli RH,McMahon CJ,Dreyer WJ,et al.Clinical characterization of left ventricular noncompaction in children:a relatively common form of cardiomyopathy[J].Circulation,2003,108(21):2672-2678.
[31]Jenni R,Oechslin E,Schneider J,et al.Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction:a step towards classification as a distinct cardiomyopathy[J].Heart,2001,86(6):666-671.
[32]Klaassen S,Probst S,Oechslin E,et al.Mutations in sarcomere protein genes in left ventricular noncompaction[J].Circulation,2008,117(22):2893-2901.
[33]Vatta M,Mohapatra B,Jimenez S,et al.Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction[J].J Am Coll Cardiol,2003,42(11):2014-2027.
[34]Brady RO,Gal AE,Bradley RM,et al.Enzymatic defect in Fabry’s disease.Ceramidetrihexosidase deficiency[J].N Engl J Med,1967,276(21):1163-1167.
[35]Lopez-Gallardo E,Lopez-Perez MJ,Montoya J,et al.CPEO and KSS differ in the percentage and location of the mtDNA deletion[J].Mitochondrion,2009,9(5):314-317.
