神经嵴发育异常导致综合征型耳聋的机制
2014-05-10刘亚兰张华冯永
刘亚兰,张华,冯永
1.中南大学湘雅医院耳鼻咽喉头颈外科,长沙 410008;
2.耳鼻咽喉重大疾病研究湖南省重点实验室,长沙 410008;
3.中国医学遗传学国家重点实验室,长沙 410078;
4.上海交通大学医学院附属仁济医院耳鼻喉科,上海 200127
耳聋是人类常见高发的致残性疾病,其中约50%的耳聋由听觉创伤、耳毒性药物聋、病毒感染、细菌感染等环境因素引起;另外 50%的先天性耳聋及临床上原因不明的后天性耳聋由遗传因素引起,称为遗传性耳聋(Hereditary hearing loss, HHL)。依据是否伴随其他组织器官症状,遗传性耳聋分为非综合征型耳聋(Nonsyndromic hearing loss, NSHL)和综合征型耳聋(Syndromic hearing loss, SHL)两类,所占比例分别为 70%和 30%[1]。几十年来,国内外的研究小组对非综合征型耳聋的遗传基础、发病机制及治疗干预进行了大量研究,取得了巨大的研究进展,大量的非综合征性耳聋致病基因被定位和克隆(已成功定位134个NSHL基因位点,克隆了63个NSHL致病基因(http://hereditaryhearingloss.org/),初步建立了相应的基因诊断技术并推广到临床。而大多数综合征型耳聋发病率相对较低,全身病变的临床表现变化多样,除了听力障碍,同时伴有眼、骨、肾、皮肤、神经系统、代谢内分泌系统等其他器官或系统的异常,种类繁多且较复杂,对病患的生存质量影响更大。目前已报道的SHL有400多种[2],大多数发病率低,临床上较常见的有 Waardenburg综合征(Waardenburg syndrome, WS)、先天性小耳畸形综合征、前庭导水管扩大综合征(Large vestibular aquduct syndrome, LVAS)等。由于SHL临床和遗传异质性极强,涉及的致病基因繁多,对其遗传基础及发病机制的系统研究变得十分困难。本课题组在前期研究WS分子机制时发现其致病基因 PAX3、SOX10及MITF之间存在相互作用关系。综合征型耳聋基因涉及多个系统,却可以导致很多共同的临床表型,表明它们之间可能存在着潜在的共同通路和相互作用网络。我们通过查阅大量文献,发现 WS和小耳畸形综合征的发病机制理论存在共性,都是由于神经嵴发育不全导致的细胞或组织的病变。基于此,本文着眼于国际耳科研究领域的前沿与难点,通过查阅大量文献并结合本研究团队前期的研究成果,首次针对神经嵴发育异常导致相关综合征型耳聋的共同通路和致病机制的研究进展进行系统地阐述,分析并归纳了与综合征型耳聋发病相关的神经嵴发育异常相关基因互作网络,为系统地研究常见综合征型耳聋致病基因的定位克隆以及发病机制提供研究思路和理论基础。
1 神经嵴细胞与神经嵴病概述
神经嵴细胞(Neural crest cells, NCC)是脊椎动物胚胎发育过程中出现的一个暂时性、多潜能细胞群,起源于背神经管的隆起——神经嵴(Neural crest,NC)。NCC形成后向外周迁移,分化为色素细胞、交感神经节的神经元细胞、神经胶质细胞、骨和软骨细胞、肾上腺髓质细胞以及平滑肌细胞等,并逐渐分化为神经嵴源性衍生物,包括颅面部、心脏、周围神经系统等多种重要的组织结构[3,4]。NCC的发育不全会导致上述细胞和组织的相关病变,统称为神经嵴病[5],包括 WS等色素异常表型相关疾病和先天性小耳畸形等颅面部畸形相关疾病,约有20余种(表 1)。
2 神经嵴异常参与WS和小耳畸形的发病机制
2.1 神经嵴发育不全理论与Waardenburg综合征
Waardenburg综合征(WS)又称听力-色素综合征,以感音神经性耳聋和皮肤、毛发、虹膜、内耳色素分布异常为主要临床表现[6]。WS是最常见的综合征型耳聋之一,人群发病率为1/42000,聋哑人群中发病率为0.9%~2.8%[7,8],占先天性耳聋的2%~5%[9,10]。WS表型的不完全外显和致病基因的多样性导致其具有高度的临床和遗传异质性[11~13]。依据不同的伴随表型,可将WS分为4种亚型(WS1-4),最常见的为 WS1和 WS2[14]。
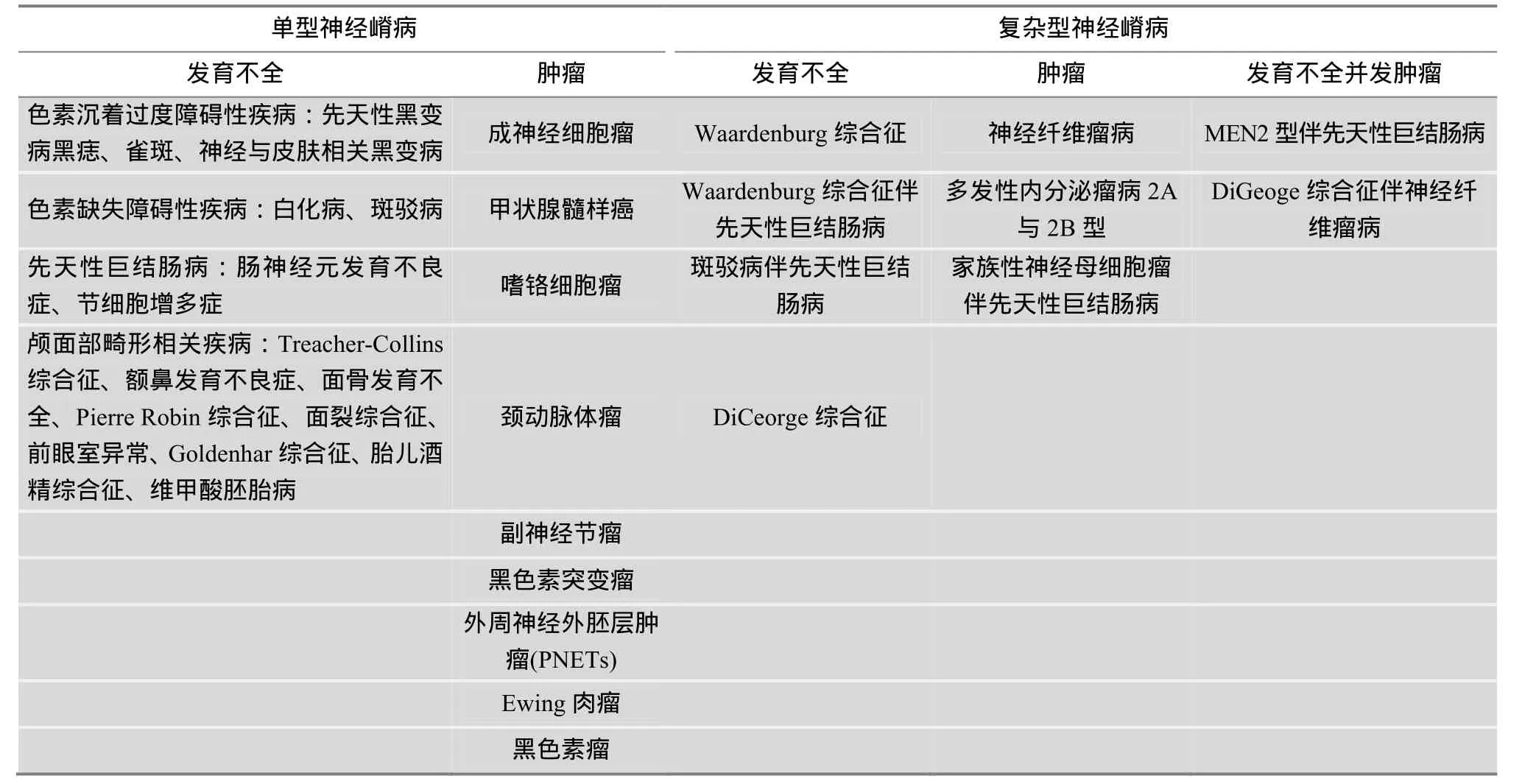
表1 已报道的神经嵴病
目前关于WS所具有的临床表型的病因有多种不同假说,其中神经嵴发育不全理论是最被认可的一种假说[15~18]。WS以黑素细胞分化缺陷发育异常导致的色素分布异常和耳聋为主要特征,黑素细胞由NC分化而来,因此WS是由于NCC的发育异常导致的一组临床症候群。NCC通过增殖、迁徙、生存、分化等过程逐渐演变为黑素前体细胞—成黑素细胞后到达真皮、表皮、内耳血管纹和眼睛的脉络膜,并逐渐分化为黑素细胞[19~21]。黑素细胞最主要的功能是产生黑色素以确保毛发和皮肤的色素沉着[22~24]。与WS相关的6个基因编码的转录因子(MITF、PAX3、SOX10和SNAI2)和信号分子(ENDRB和EDN3)均参与这一过程,在NC的发育中发挥重要的调控作用。不同的致病基因与不同的WS亚型相关联,PAX3基因突变可导致WS1和 WS3[25,26],MITF基因突变导致WS2A[27],SNAI2基因突变导致 WS2D[28],SOX10突变导致 WS2E[29,30]。SOX10、EDN3和 EDNRB基因突变可造成WS4[31~33]。国外已有的WS突变数据库报道WS有近280个基因突变位点(http://grenada.lumc.nl/LOVD2/WS/,截至2014年6月);而国内相关研究[34~41]表明中国人群 WS主要致病基因为PAX3、MITF和SOX10,已发现相关突变位点23种,目前尚无SNAI2、EDN3和EDNRB基因突变及WS3病例报道。上述WS相关基因的突变会造成NC发育异常,导致黑素细胞发育不良,引起黑色素合成减少,表现出皮肤、毛发、虹膜低色素等临床症状。血管纹是内耳耳蜗结构之一,在内淋巴生成过程中起重要作用。研究显示,黑素细胞发育不良会引起血管纹中黑素细胞源性的中间细胞缺乏进而造成柯蒂氏器(Corti)退化变性,最终导致感音神经性耳聋[42]。此外,由于颅面骨、肠壁神经节以及四肢骨骼肌肉等均为神经嵴源性衍生物,因此 NC的发育异常必然会导致这些组织和器官的发育异常,从而产生WS的一系列伴随症状。
2.2 神经嵴细胞迁徙理论与小耳畸形
先天性小耳畸形综合征也是常见的一类综合征型耳聋。小耳畸形(或称为先天性外中耳畸形)表现为重度耳廓发育不全,有外耳道闭锁或狭窄,中耳甚至内耳畸形,通常伴有听力损失,需要耳再造术治疗[43]。小耳畸形可以作为一个独立的出生缺陷或作为综合征的一部分[44]。流行病学调查显示,全球小耳无耳畸形总发病率为 2.06/10000,小耳畸形为1.55/10000,无耳畸形为0.36/10000。由于入选标准的差异,不同地区的流行病学调查结果具有一定差异。意大利、法国、瑞典、芬兰和美国的流行病学调查结果显示,小耳畸形的发病率范围为 0.83~4.34/10000,而西班牙、亚洲、美洲印第安和安第斯的发病率被认为更高[43,45~49]。在我国,研究显示先天性小耳畸形的发病率约为 1.40/10000,其中无耳为0.55/10000,小耳为0.85/10000,城镇发病率高于农村。全国以新疆发病率最高,为 2.08/10000。男女发病比例约为 2:1,右侧畸形较多,双侧者约10%[50,51]。目前,国内外已经报道的小耳畸形相关综合征大约有 20余种,其中常见的有 Treacher Collins综合征、Branchiooto-Renal综合征、Goldenhar综合征、Nager综合征及Miller综合征等。相关研究已鉴定的小耳畸形相关综合征的致病基因大约有 20余个(表 2)。
目前对先天性小耳畸形的病因和表型变异性还知之甚少,然而,发育生物学和遗传学研究表明,神经嵴细胞功能缺陷与小耳畸形等多种颅面综合征相关。Luquetti等[43]认为神经嵴细胞紊乱和血管破坏可能为小耳畸形的病因。NCC可分成颅神经嵴细胞(Cranial neural crest cell, CNCC)、心神经嵴细胞(Cardiac neural crest cells)、迷走神经嵴细胞(Vagal neural crest cells)以及体神经嵴细胞(Trunk neural crest cells)4个主要的轴向细胞群,每一部分都沿特定路径迁移并分化成特定的细胞[52]。其中,颅神经嵴细胞在耳部发育中起重要作用。颅神经嵴细胞是起源于背侧神经管的一个暂时性、可迁移、多潜能的细胞群,可以进一步分化为前脑(Forebrain)、中脑(Midbrain)、后脑(Hindbrain),其中,后脑的CNCC进一步分区形成数个菱脑原节(Rhombomere′r),这一结构在颅颌面部的发育中起重要作用[53]。菱脑原节不仅为 CNCC 形成数支彼此分隔的迁移流提供了解剖结构基础,而且防止了颅颌面部神经和骨等结构的发育发生位置错乱。CNCC在迁移时,不是以整体的细胞团形式进行迁移,而是被“NCC缺失区”分割成若干特定的细胞支流,在各种微环境的诱导下,沿着特定的精确路线迁移至颌面部的特定部位,进一步分化为颅颌面部的软硬组织结构(图 1)。其中,耳廓起源于胚胎第一鳃弓(下颌弓)和第二鳃弓(舌骨弓)[54]。在耳廓的发育阶段,胚胎受到遗传或外界因素影响,容易出现耳廓的多种发育畸形。小耳畸形就是源于胚胎期第一、二鳃弓、第一咽囊、第一鳃裂和颞骨原基的颅骨结构发育不良的一组先天性畸形。研究发现,在小鼠中耳廓发育期镫骨动脉破裂会引起小耳畸形,使CNCC不具有迁移性[55],而CNCC能否正确迁移到指定位置,是颅颌面结构正常发育的前提,也是各种颌面部畸形发生的重要原因。
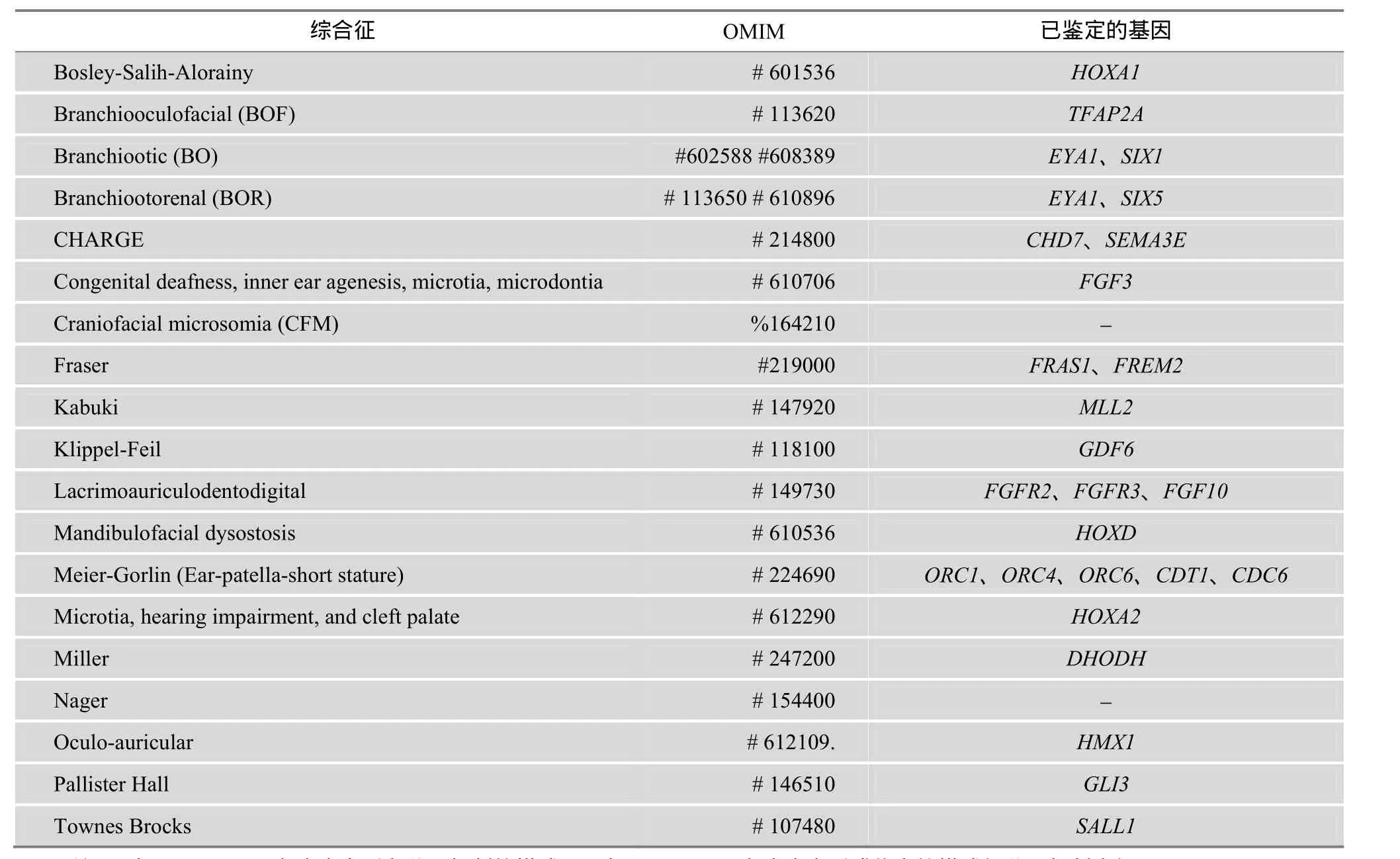
表2 已经鉴定的小耳畸形相关综合征致病基因信息

图1 神经嵴细胞分化发育至耳的过程示意图
3 以PAX3和SOX10为中心的神经嵴迁移分化相关基因互作网络与SHL
神经嵴细胞功能异常不仅能导致神经嵴起源的黑素细胞缺失而造成的WS,还与包括小耳畸形在内的多种颅面综合征相关,表明 WS和小耳畸形综合征的发病机制理论存在共性。本课题组前期对 WS分子机制进行研究时发现,其致病基因PAX3、SOX10及 MITF之间存在相互作用关系[56,57];以此理论为依据,通过查阅大量相关文献发现有多种基因和细胞因子共同参与神经嵴细胞不同阶段迁移的调控,与神经嵴发展相关的基因(如PAX3、SOX10、MITF、HOX、EGR2、EYA1、SALL1和SIX1等)相互之间存在一定联系,并参与WS等色素异常表型相关SHL和小耳畸形表型相关综合征的致病机制。
3.1 SOX10与PAX3基因在神经嵴细胞迁移分化中的作用
SOX10与 PAX3基因编码的蛋白不仅是具有特殊结构的转录因子,还是起正调控作用的反式作用因子,通过DNA结合域直接或间接结合靶基因的上游启动子顺时作用元件来调控靶基因表达。缺失DNA结合域的SOX10与PAX3突变蛋白由于不能与DNA结合,导致靶基因转录水平下降而不表达。研究表明,SOX10与 PAX3基因在神经嵴细胞的迁移和分化中起到了十分重要的作用[58]。
配对盒基因(Pair box 3,PAX3)位于染色体2q35-2q37,编码的蛋白是高度保守的转录因子,由479个氨基酸组成,含有 4个高度保守的结构域,分别为配对盒结构域(Paired box domain, PD)、同源结构域(Homeodomain, HD)、锌肽序列和转录激活域(TA)。其中 PD在 PAX3的结构和功能中起重要作用,PD通过与SOX10的HMG相互作用以调控PAX3活性[59],而且PAX3必须通过PD与SOX10结合才能协同激活c-RET和MITF的转录[60,61]。此外,大多数PAX3突变都发生于含有部分PD结构域的2号外显子上,这些无义或移码突变由于影响了PD结构域与DNA的紧密结合而导致PAX3的功能异常。PAX3突变可导致心脏神经嵴功能异常、神经嵴源性细胞组织功能不良、神经管畸形、中枢神经系统异常以及四肢肌肉发育不良等多种组织病变。人的PAX3杂合突变可导致轻度的WS1表型,而复合杂合或纯合PAX3突变则导致严重的WS3表型[62]。此外,PAX3突变还能引起颅面-耳聋-手综合征(Craniofacial-deafness-hand syndrome, CDHS,MIM:122880)[63]。PAX3不仅在维持干细胞的多潜能性、细胞系迁移、分化增殖、凋亡和抑制终末分化中起重要作用[18],而且在胚胎的神经嵴及其衍生物的发育中起关键作用。Tremblay等[64]用转基因神经解剖标记 PAX3在Splotch小鼠的神经嵴发育中的作用,表明PAX3在神经嵴源性结构特别是颅神经节和神经的发育中起重要作用。神经嵴细胞中PAX3的降低将导致神经嵴细胞过早凋亡,引起相关发育疾病。Wu等[65]发现在神经嵴中 PAX3的持续表达会导致小鼠出现腭裂和缺陷成骨,该研究为PAX3的下调表达在多能神经嵴前体分化和颅脑发育过程中起到的重要作用提供了体内证据。人和小鼠PAX基因表达水平下调影响神经嵴的正常发育,说明神经嵴的发育依赖 PAX3的正常功能,PAX3突变可导致多种神经嵴源性细胞的功能异常。目前,PAX3调控神经嵴发育的具体机制尚不明确,但在神经嵴的发育过程中,PAX3通过调控编码TYR受体的c-RET、信号分子Wnt1和转化生长因子 β2(TGF-β2)等下游靶基因的表达促进神经嵴的迁移、分化和增殖[66~68]。c-RET、Wnt1和TGF-β2是神经嵴发育的关键调控因子。c-RET酪氨酸激酶受体跨越神经嵴源性细胞胞膜,是细胞的动力生存和分化增殖所必需的。PAX3在神经嵴源性细胞中通过调节c-RET的表达刺激肠道神经节的正常迁移和发育[69]。PAX3直接与c-RET启动子上的增强子元件结合后协同SOX10激活c-RET表达,这种协同方式受染色质乙酰化调节。Wnt1是PAX3的下游基因,缺乏PAX3的小鼠胚胎背神经管中的Wnt1表达减少[70]。神经嵴细胞分化受PAX3和ZIC1的协同调控,Wnt可使神经嵴细胞迁移前PAX3的表达增加,迁移后 PAX3表达下降,而 Wnt对神经嵴细胞的这种作用可被骨形成蛋白4阻断[71]。TGF-β2是调节细胞行为和影响细胞动力、增殖分化和凋亡的关键因子。许多神经嵴源性细胞十分依赖TGF-β2,研究表明TGF-β2纯合突变小鼠可导致神经嵴病,影响颅面骨结构、耳、心脏和泌尿生殖系统。缺乏PAX3表达的突变小鼠胚胎与野生胚胎相比,TGF-β2表达明显减少[72]。此外,在神经嵴迁移早期,PAX3与FoxD3的比例还决定神经嵴细胞向神经/神经胶质细胞或是色素细胞转变[73]。
性别决定区盒基因(SRY(sex determining region Y)-box 10, SOX10)位于染色体22q13.1,编码的蛋白含有466个氨基酸,是高度保守的转录因子,由高活性组分结构域(High mobility group, HMG)、Group E结构域和C端转录激活域(TA)构成,其中HMG域的主要功能是识别并结合DNA[74]。酵母双杂交分析结果表明,SOX10通过HMG域的N端直接与KROX-20、PAX3、PAX6、HOXA3等至少17种可调节SOX10转录活性的蛋白相互作用[75]。SOX10为神经嵴分化和少突细胞分化所必需,人的杂合SOX10突变会导致一些神经嵴源性病变和脑白质病变,相关疾病表型包括WS2和WS4[29],外周和中枢髓鞘形成障碍,PCWH(OMIM:609136)[76]、HSCR(OMIM:142623)[77]和YDBS(OMIM:601706)[78]等。SOX10基因突变绝大部分为无义突变和移码突变,导致终止密码子提前出现并产生截短蛋白而致病[79],此外,修饰基因的活性、突变导致的单倍体剂量不足以及其他原因引起的SOX10蛋白功能改变可部分解释上述疾病表型的致病原因[80]。SOX10最先在NCC迁移早期的背神经管中表达,然后随着NCC逐渐分化而在周围神经系统、ENS和黑素细胞等衍生物如中表达。在人和成年小鼠中广泛表达于大脑、心脏、肺、肾上腺、结肠、膀胱、胰腺、前列腺、睾丸、内耳等各种组织器官中[81]。SOX10是神经嵴源性细胞迁移分化的一种关键转录因子,可单独或协同其他转录因子,通过与 MITF、DCT、TYR、TYRP1、RET、MPZ、GJB1和EDNRB等下游靶基因的启动子或增强子结合直接或间接参与黑色素合成。SOX10在黑素细胞和肠道神经节的发育中发挥重要作用,可促进胚胎神经细胞和外周神经系统的发育,不仅为胚胎发育早期所必需,而且对胚胎后期的小肠神经系统的发育也非常重要[82]。而SOX10在内耳发育早期广泛表达[83,84],提示SOX10突变导致的耳聋可能还有另外的机制。SOX10基因敲除小鼠模型的研究表明前体感觉祖细胞的表达减少会导致耳蜗变短[85]。这一观察结果与SOX10突变耳聋患者内耳MRI/CT扫描发现的形态学异常结果相同[85,86]。Mao等[87]利用神经脊特异转录因子SOX10缺失小鼠模型来评估Schwann细胞缺陷在内耳神经支配中的作用,发现神经脊来源的Schwann细胞为螺旋神经节神经元的迁移提供了终止信号,并能通过螺旋神经节传导促进耳蜗Corti器的正确定位。
3.2 PAX3-SOX10-MITF通路参与色素异常表型相关的综合征型耳聋的发病机制
PAX3-SOX10-MITF通路与色素异常相关耳聋综合征的发病机制有重要联系[88]。神经嵴及其起源的组织细胞的发育过程复杂且受多个转录因子、信号通路和生长因子的调控[89]。WS致病基因MITF、PAX3、SOX10和SNAI2编码的转录因子以及EDN3和EDNRB编码的信号传导分子都参与其中并发挥重要作用[90]。这些转录因子的调控作用与 WS等色素异常表型相关疾病的病理生理明显相关,他们之间的相互作用以及他们与其他基因之间的相互作用构成复杂的调控网络调控 NCC源性细胞和组织尤其是黑素细胞的发育,表现为以 MITF为中心的调控与被调控的功能性联系[91~93]。WS 致病基因 MITF、PAX3、SOX10之间存在相互作用。研究显示SOX10与PAX3可单独或协同激活并上调 MITF 表达[94]。SOX10通过与MITF启动子的高度保守序列结合后使MITF转录活性提高 100倍,且 PAX3可增强这种作用。SOX10与 MITF协同激活负责黑色素合成的多巴色素互变异构酶(Dopachrome tautomerase, DCT),而PAX3拮抗MITF对DCT的转录[95]。同时MITF可调控SNAI2表达[28],SOX10调控MITF对TYR表达的诱导和整个黑素细胞的分化[96]。SOX10在ENS和黑素细胞发育中都能够调控EDNRB的表达[97],而EDNRB又可通过信号传导通路调控MITF转录[98](图2,④⑤⑧PAX3-MITF-SOX10、MITF--EDNRB 通路)。本研究小组前期研究[56,57]表明,PAX3、SOX10与 MITF突变通过单倍体剂量不足分别导致WS1和WS2,突变蛋白可与其自身或正常的野生蛋白结合形成二聚体,使野生蛋白与靶基因 DNA不能正确结合而失去调节黑色素细胞靶基因的活性。此外,PAX3和SOX10基因突变可通过显性负效应抑制野生 PAX3和SOX10蛋白功能而导致严重的WS3和WS4。
除了 WS,PAX3-SOX10-MITF通路还可能与Leopard综合征及Noonan综合征等色素异常表型相关耳聋综合征的发病机制有关。Leopard综合征是一类以多发性黑斑为主要临床表现的色素异常型综合征,有15%~25%的患者出现感音神经性耳聋,其最常见的致病基因是 PTPN11基因[99]。研究表明,PTPN11在 MET/Gab1复合物特殊信号中起重要作用,Gab1作为酪氨酸激酶受体c-MET的基质,通过直接结合c-MET的结合域,参与c-MET的特异性分化。Gab1突变的过表达通过与PTPN11相互作用阻止HGF/SF诱导的MAPK通路的激活,从而在c-MET/Gab1的特殊信号中发挥重要作用[100](图2,⑦ METPTPN11通路)。而肝细胞生长因子(HGF)/ c-MET的信号被认为在黑素细胞的发展和黑色素瘤转移中起关键作用。PAX3、SOX10以及MITF通过调节MET基因的表达在黑色素瘤细胞中起重要作用[101];MITF通过上调MET的表达帮助肝细胞生长因子保护黑色素细胞并调控黑色素细胞瘤凋亡[102]。PAX3和MITF以及MET的启动子独立结合,可以独立调控并激活MET,而SOX10不能直接激活MET,但是可以通过协同 MITF和 PAX3激活 MET[101](图 2,⑥ METMITF通路)。除了Leopard综合征,PTPN11基因的突变还能在大约50%的Noonan综合征病例中检出。此外,RAF1基因的杂合突变也能同时在Leopard综合征及Noonan综合征患者中检出[103]。
综上所述,这些色素相关型耳聋综合征除了共同具有色素发育异常和神经性耳聋的临床表型,还存在共同的致病基因。这些基因可能通过他们之间的相互作用以及他们与其他基因之间的相互作用构成复杂的调控网络调控 NCC 源性细胞和组织尤其是黑素细胞的发育,从而在伴有色素异常表型的综合征型耳聋的发病机制中发挥作用。
3.3 以PAX3和SOX10为中心的基因互作通路参与小耳畸形表型相关综合征的发病机制
SOX10和PAX3,以及通路中涉及的EYA1、SIX1、SALL1、HOX基因家族、EGR2(KROX-20)以及FGF3等基因与小耳畸形表型相关综合征的发病机制有重要联系。耳腮肾综合征(Branchio-oto-renal syndrome,BOR)和Townes Brocks综合征是小耳畸形相关综合征里相对多见的两个综合征,二者都有混合型耳聋和小耳畸形的临床表型[104,105]。EYA1和SALL1分别是这两种综合征的致病基因,与PAX家族和SIX1基因联系密切[106,107]。在内耳中,反式激活磷酸酶 EYA1与同源蛋白SIX1交互形成转录激活复合物,调节感应和神经细胞的增殖、存活以及内耳发育过程中的分化诱导[108]。SALL1是EYA/SIX复合体的下游调控靶基因。SIX1直接结合SALL1的启动子并以剂量依赖性的方式诱导SALL1表达。而SIX1与PAX、EYA1基因及其产物在复杂的网络中具有相互依存关系,形成正、负反馈回路,在肾脏发育过程中具有一定的网络作用[109](图 2,①②③ PAX-EYA-SIX-SALL1通路)。Fatemeh等[110]对来自伊朗的一个家系的小耳畸形患者的DNA序列进行分析,结果发现一个HOXA2基因的同源结构变异(p.Q186K)是该患者发生小耳畸形的重要原因。Hox基因家族编码高度保守的转录因子,参与腮弓的发育。HOXA1基因失活导致小鼠外耳、中耳和内耳的发育不良。而HOXA1/HOXB1复合物的突变则会导致无耳。而HOXA2可能作用于定义第二腮弓的特性以及耳廓形成的初始阶段,在小鼠耳廓高表达。HOXB6和HOXA7基因缺失的小鼠表达出小耳畸形并伴有眼裂增宽,唇腭裂。Hox基因家族在各菱脑的特殊表达通过 EGR2的直接调节以控制后脑的分节[111,112](图 2,⑩ EGR2-HOXA2通路)。SOX10作为神经嵴细胞分化迁移中重要的转录因子之一,和EGR2在神经嵴细胞迁移中共表达。在自激活过程中,SOX10是EGR2在神经嵴细胞中自动调节重要的共活化剂[113]。Reiprich等[113]研究表明,在雪旺氏细胞中,SOX10影响髓鞘形成,通过直接激活髓鞘基因和诱导 EGR2调节外围髓鞘的形成。SOX10与转录因子(POU3F1和POU3F2)协同作用激活EGR2后,再协同EGR2激活下游靶基因。这种依赖SOX10转录的连锁模式在雪旺氏细胞和黑素细胞中一样(图 2,⑨ SOX10-EGR2通路)。伴有小耳畸形表型的 LAMM 综合征目前已知的唯一致病基因是FGF3,研究表明LAMM综合征与FGF3的隐性突变共分离[114]。FGF3与包括SOX10在内的至少5个基因家族的调节基因直接或间接受到EGR2在神经嵴细胞迁徙时r3/r5的控制,FGF3是后脑中控制EGR2表达的重要信号分子,抑制 FGF3的信号会下调EGR2的表达[115](图2,⑪FGF3-EGR2通路)。
综上所述,本文通过查阅大量文献并结合本课题组前期的研究基础,以SOX10和PAX3基因为中心,总结并绘制了与神经嵴细胞的迁移和分化有关的基因相互作用网络以及该通路涉及的多种综合征型耳聋的关系图(图2)。
3.4 神经嵴细胞迁移分化相关基因互作网络涉及前庭导水管扩大表型的致病机制
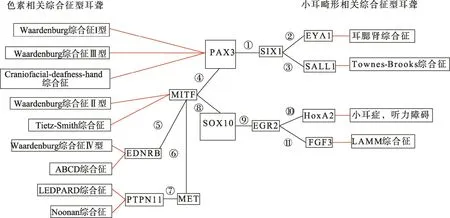
图2 与神经嵴细胞的迁移和分化有关的基因相互作用网络及涉及的综合征
值得注意的是,在上述与神经嵴发育异常相关的综合征型耳聋中,也存在不少合并有前庭导水管扩大的内耳畸形的报道。Madden等[116]通过对 9个Warrdenburg综合征患儿和正常儿童进行统计学分析比较,发现所有患者均有听力损失,50%的患儿出现前庭导水管中段扩大。Elmaleh-Bergès等[117]对收集到的由SOX10导致的Waardenburg综合征患者进行内耳影像学观察,发现双侧内耳异常(包括半规管的缺如和发育不全,前庭导水管扩大以及耳蜗畸形)极有可能与Waardburg综合征的致病基因SOX10有关。耳腮肾综合征患者也经常伴有前庭导水管扩大表型。Stinckens等[118]对一个耳腮肾综合征大家系进行了研究,结果显示部分患者伴有扩大的前庭导水管和渐进性感音神经性耳聋。Kemperman等[119]对两名耳腮肾综合征患者(父子)进行长期听力随访检测以及岩骨 CT成像研究,发现进行波动性听力损失和热量功能减退与前庭导水管扩大相关。Propst等[120]通过 CT断层扫描将耳腮肾综合征的患者和正常人群进行比较,结果发现,耳腮肾综合征患者内耳CT表现和对照组之前存在明显差异(30/39,76.9%),最常见的特征性表现有:耳蜗顶圈发育不良、面神经偏向耳蜗内侧、漏斗状内听道、咽鼓管异常开放。鉴于前庭导水管扩大在耳腮肾综合征中出现较多,Ito等[121]以 SIX1基因突变的耳腮肾综合征的患者为研究对象,探寻SIX1基因突变和前庭导水管扩大之前的关系;发现在SIX1的Y129C突变可能导致前庭导水管扩大和耳腮肾综合征的发生。以上研究提示:与神经嵴细胞的迁移和分化有关的基因相互作用网络可能在 WS等色素异常以及小耳畸形相关的耳聋综合征的发病机制中起重要作用,还可能参与前庭导水管扩大表型的致病机制。
4 结语与展望
以往耳聋研究往往局限于对单个基因突变致病机理进行研究。而如今有许多临床表型无已法用单基因突变进行解释。在机体内,基因编码产物往往不是单独发挥作用,而是形成较大的复合体,在特定时间和空间行使特定的功能,所以基因功能的研究应突破单基因研究模式,主张研究基因间的相互作用,最终在一个复杂的基因网络中全面认识基因功能。综合征型耳聋基因涉及多个系统,却可以导致很多共同的临床表型,这表明它们之间可能存在着潜在的共同通路和相互作用网络。其中某一环节出现功能障碍即有可能影响网络的整体功能。这方面的研究有过成功的例子,Usher综合征中多种USH1蛋白(Cadherin23、Harmnin、Protocadherin和Myosin VIIa)之间存在着复杂的相互作用关系已经得到证实。Brodbeck等[111]的研究提出理论,由于进化是保守的,基因相互作用的网络在不同物种的不同器官中可能存在共同的作用机制和通路。以PAXEYA-SIX调节通路为例,小鼠的遗传和生化研究表明,SIX1与PAX、EYA1在肾脏发育过程中有一定的网络作用,这些基因和它们各自的产物作用在复杂的网络中具有相互依存关系,形成正负反馈回路。鉴于在果蝇中也有同样的阶梯效应,推断PAX-EYA-SIX调节通路广泛存在于哺乳动物肾脏发育过程中。这种相互作用关系在果蝇和小鼠中有同样的效应,那么在耳发育过程中也可能存在相似的网络效应。目前国内外的综合征型耳聋相关研究分散,规模小,人群样本量小且病例资源有限,鉴定的基因种类和数量严重不足,而且即便是已鉴定的致病基因对其致聋机制的研究亦不系统和深入,导致尚有绝大部分综合征型耳聋患者不能得到明确的基因诊断和临床治疗。Waardenburg综合征、先天性小耳畸形相关综合征以及前庭导水管扩大表型等SHL的发病机制存在共性,都与神经嵴细胞迁移分化相关的基因互作网络有关,未来针对综合征型耳聋潜在的共同通路和致病机制进行系统地研究,可为综合征型耳聋的研究提供新的思路和理论基础。
[1]Van Camp G, Willems PJ, Smith RJ.Nonsyndromic hearing impairment: unparalleled heterogeneity.Am J Hum Genet, 1997, 60(4): 758–764.
[2]Schrijver I, Gardner P.Hereditary sensorineural hearing loss: advances in molecular genetics and mutation analysis.Expert Rev Mol Diagn, 2006, 6(3): 375–386.
[3]Chai Y, Jiang X, Ito Y, Bringas P Jr, Han J, Rowitch DH,Soriano P, McMahon AP, Sucov HM.Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis.Development, 2000, 127(8): 1671–1679.
[4]Basch ML, Bronner-Fraser M.Neural crest inducing signals.Adv Exp Med Biol, 2006, 589: 24–31.
[5]Bolande RP.Neurocristopathy: its growth and development in 20 years.Pediatr Pathol Lab Med, 1997,17(1): 1–25.
[6]Waardenburg PJ.A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness.Am J Hum Genet, 1951, 3(3):195–253.
[7]Farrer LA, Grundfast KM, Amos J, Arnos KS, Asher JH Jr, Beighton P, Diehl SR, Fex J, Foy C, Friedman TB,Greenberg J, Hoth C, Marazita M, Milunsky A, Morell R, Nance W, Newton V, Ramesar R, Agustin TB, Skare J, Stevens CA,Wagner RG, Wilcox ER, Winship I, Read AP.Waardenburg syndrome (WS) type I is caused by defects at multiple loci, one of which is near ALPP on chromosome 2: first report of the WS consortium.Am J Hum Genet, 1992, 50(5): 902–913.
[8]Reed WB, Stone VM, Boder E, Ziprkowski L.Pigmentary disorders in association with congenital deafness.Arch Dermatol, 1967, 95(2): 176–186.
[9]Hodgkinson CA, Nakayama A, Li H, Swenson LB, Opdecamp K, Asher JH Jr, Arnheiter H, Glaser T.Mutation at the anophthalmic white locus in Syrian hamsters:haploinsufficiency in the Mitf gene mimics human Waardenburg syndrome type 2.Hum Mol Genet,1998, 7(4): 703–708.
[10]Read AP, Newton VE.Waardenburg syndrome.J Med Genet, 1997, 34(8): 656–665.
[11]Arias S.Genetic heterogeneity in the Waardenburg syndrome.Birth Defects Orig Artic Ser, 1971, 7(4):87–101.
[12]Hageman MJ, Delleman JW.Heterogeneity in Waardenburg syndrome.Am J Hum Genet, 1977, 29(5):468–485.
[13]Farrer LA, Arnos KS, Asher JH Jr, Baldwin CT, Diehl SR, Friedman TB, Greenberg J, Grundfast KM, Hoth C,Lalwani AK, Landa B, Leverton K, Milunsky A, Morell R, Nance WE, Newton V, Ramesar R, Rao VS, Reynolds JE, Agustin TBS, Wilcox ER, Winship I, Read AP.Locus heterogeneity for Waardenburg syndrome is predictive of clinical subtypes.Am J Hum Genet,1994, 55(4): 728–737.
[14]Liu XZ, Newton VE, Read AP.Waardenburg syndrome type II: phenotypic findings and diagnostic criteria.Am J Med Genet, 1995, 55(1): 95–100.
[15]Mollaaghababa R, Pavan WJ.The importance of having your SOX on: role of SOX10 in the development of neural crest-derived melanocytes and glia.Oncogene,2003, 22(20): 3024–3034.
[16]Baxter LL, Hou L, Loftus SK, Pavan WJ.Spotlight on spotted mice: a review of white spotting mouse mutants and associated human pigmentation disorders.Pigment Cell Res, 2004, 17(3): 215–224.
[17]Spritz RA, Chiang PW, Oiso N, Alkhateeb A.Human and mouse disorders of pigmentation.Curr Opin Genet Dev, 2003, 13(3): 284–289.
[18]Pingault V, Ente D, Dastot-Le Moal F, Goossens M,Marlin S, Bondurand N.Review and update of mutations causing Waardenburg syndrome.Hum Mutat, 2010,31(4): 391–406.
[19]Crane JF, Trainor PA.Neural crest stem and progenitor cells.Annu Rev Cell Dev Biol, 2006, 22: 267–286.
[20]Dupin E, Le Douarin NM.Development of melanocyte precursors from the vertebrate neural crest.Oncogene,2003, 22(20): 3016–3023.
[21]Sauka-Spengler T, Bronner-Fraser M.Development and evolution of the migratory neural crest: a gene regulatory perspective.Curr Opin Genet Dev, 2006, 16(4):360–366.
[22]Brito FC, Kos L.Timeline and distribution of melanocyte precursors in the mouse heart.Pigment Cell Melanoma Res, 2008, 21(4): 464–470.
[23]Steingrímsson E, Copeland NG, Jenkins NA.Melanocytes and the microphthalmia transcription factor network.Annu Rev Genet, 2004, 38: 365–411.
[24]Yajima I, Larue L.The location of heart melanocytes is specified and the level of pigmentation in the heart may correlate with coat color.Pigment Cell Melanoma Res, 2008, 21(4): 471–476.
[25]Tassabehji M, Read AP, Newton VE, Harris R, Balling R, Gruss P, Strachan T.Waardenburg's syndrome patients have mutations in the human homologue of the Pax-3 paired box gene.Nature, 1992, 355(6361):635–636.
[26]Hoth CF, Milunsky A, Lipsky N, Sheffer R, Clarren SK,Baldwin CT.Mutations in the paired domain of the human PAX3 gene cause Klein-Waardenburg syndrome(WS-III) as well as Waardenburg syndrome type I(WS-I).Am J Hum Genet, 1993, 52(3): 455–462.
[27]Tassabehji M, Newton VE, Read AP.Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene.Nat Genet, 1994, 8(3):251–255.
[28]Sánchez-Martín M, Rodríguez-García A, Pérez-Losada J,Sagrera A, Read AP, Sánchez-García I.SLUG (SNAI2)deletions in patients with Waardenburg disease.Hum Mol Genet, 2002, 11(25): 3231–3236.
[29]Bondurand N, Dastot-Le Moal F, Stanchina L, Collot N,Baral V, Marlin S, Attie-Bitach T, Giurgea I, Skopinski L, Reardon W, Toutain A, Sarda P, Echaieb A,Lackmy-Port-Lis M, Touraine R, Amiel J, Goossens M,Pingault V.Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4.Am J Hum Genet,2007, 81(6): 1169–1185.
[30]Iso M, Fukami M, Horikawa R, Azuma N, Kawashiro N,Ogata T.SOX10 mutation in Waardenburg syndrome type II.Am J Med Genet A, 2008, 146A(16): 2162–2163.
[31]Edery P, Attie T, Amiel J, Pelet A, Eng C, Hofstra RM,Martelli H, Bidaud C, Munnich A, Lyonnet S.Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome).Nat Genet,1996, 12(4): 442–444.
[32]Puffenberger EG, Hosoda K, Washington SS, Nakao K,deWit D, Yanagisawa M, Chakravarti A.A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung's disease.Cell, 1994, 79(7): 1257–1266.
[33]Pingault V, Bondurand N, Kuhlbrodt K, Goerich DE,Préhu MO, Puliti A, Herbarth B, Hermans-Borgmeyer I,Legius E, Matthijs G, Amiel J, Lyonnet S, Ceccherini I,Romeo G, Smith JC, Read AP, Wegner M, Goossens M.SOX10 mutations in patients with Waardenburg-Hirschsprung disease.Nat Genet, 1998, 18(2): 171–173.
[34]Jiang L, Chen HS, Jiang W, Hu ZM, Mei LY, Xue JJ, He CF, Liu YL, Xia K, Feng Y.Novel mutations in the SOX10 gene in the first two Chinese cases of type IV Waardenburg syndrome.Biochem Biophys Res Commun,2011, 408(4): 620–624.
[35]Yang SZ, Cao JY, Zhang RN, Liu LX, Liu X, Zhang X,Kang DY, Li M, Han DY, Yuan HJ, Yang WY.Nonsense mutations in the PAX3 gene cause Waardenburg syndrome type I in two Chinese patients.Chin Med J (Engl),2007, 120(1): 46–49.
[36]Qin W, Shu A, Qian X, Gao J, Xing Q, Zhang J, Zheng Y, Li X, Li S, Feng G, He L.A novel mutation of PAX3 in a Chinese family with Waardenburg syndrome.Mol Vis, 2006, 12: 1001–1008.
[37]Chang T, Hashimoto K, Bawle EV.Spontaneous contraction of leukodermic patches in Waardenburg syndrome.J Dermatol, 1993, 20(11): 707–711.
[38]Mancini AJ.Waardenburg syndrome type II in a Taiwanese woman with a family history of pseudoxanthoma elasticum.Int J Dermatol, 1997, 36(12): 933–935.
[39]Wang J, Li SQ, Xiao XS, Wang PF, Guo XM, Zhang QJ.PAX3 mutations and clinical characteristics in Chinese patients with Waardenburg syndrome type 1.Mol Vis,2010, 16: 1146–1153.
[40]Chen HS, Jiang L, Xie ZG, Mei LY, He CF, Hu ZM, Xia K, Feng Y.Novel mutations of PAX3, MITF, and SOX10 genes in Chinese patients with type I or type II Waardenburg syndrome.Biochem Biophys Res Commun,2010, 397(1): 70–74.
[41]Chen J, Yang SZ, Liu J, Han B, Wang GJ, Zhang X,Kang DY, Dai P, Young WY, Yuan HJ.Mutation screening of MITF gene in patients with Waardenburg syndrome type 2.Hereditas, 2008, 30(4): 433–438.
[42]Tachibana M, Kobayashi Y, Matsushima Y.Mouse models for four types of Waardenburg syndrome.Pigment Cell Res, 2003, 16(5): 448–454.
[43]Luquetti DV, Heike CL, Hing AV, Cunningham ML,Cox TC.Microtia: epidemiology and genetics.Am J Med Genet A, 2012, 158A(1): 124–139.
[44]Alasti F, Van Camp G.Genetics of microtia and associated syndromes.J Med Genet, 2009, 46(6): 361–369.
[45]Canfield MA, Langlois PH, Nguyen LM, Scheuerle AE.Epidemiologic features and clinical subgroups of anotia/microtia in Texas.Birth Defects Res A Clin Mol Teratol, 2009, 85(11): 905–913.
[46]Suutarla S, Rautio J, Ritvanen A, Ala-Mello S, Jero J,Klockars T.Microtia in Finland: comparison of characteristics in different populations.Int J Pediatr Otorhinolaryngol, 2007, 71(8): 1211–1217.
[47]Forrester MB, Merz RD.Descriptive epidemiology of anotia and microtia, Hawaii, 1986–2002.Congenit Anom (Kyoto), 2005, 45(4): 119–124.
[48]Shaw GM, Carmichael SL, Kaidarova Z, Harris JA.Epidemiologic characteristics of anotia and microtia in California, 1989–1997.Birth Defects Res A Clin Mol Teratol, 2004, 70(7): 472–475.
[49]Harris J, Källén B, Robert E.The epidemiology of anotia and microtia.J Med Genet, 1996, 33(10): 809–813.
[50]朱军, 王艳萍, 梁娟, 周光萱.1988~1992年全国先天性无耳和小耳畸形发病率的抽样调查.中华耳鼻咽喉科杂志, 2000, 35(1): 62–65.
[51]李文芳.先天性小耳畸形在家族中发病聚集报道(附2个家族4例报告).江西医药, 2008, 43(7): 708–709.
[52]Trainor PA.Specification of neural crest cell formation and migration in mouse embryos.Semin Cell Dev Biol,2005, 16(6): 683–693.
[53]卢境婷, 王旭东, 代杰文, 孙昊, 沈国芳.颅神经嵴细胞的迁移及特性.中华口腔医学研究杂志: 电子版,2011, 5(6): 652–657.
[54]Schoenwolf GC, Bleyl SB, Brauer PR, Francis-West PH.Development of the ears and eyes.Larsen’s human embryology (4th ed.), New York: Churchill Livingstone,2009.
[55]魏建军, 潘博, 于晓波, 刘磊, 赵延勇, 林琳, 庄洪兴,蒋海越.小耳畸形的“二期法”耳廓再造术.组织工程与重建外科杂志, 2010, 6(5): 276–278.
[56]Zhang H, Chen HS, Luo HJ, An J, Sun L, Mei LY, He CF, Jiang L, Jiang W, Xia K, Li JD, Feng Y.Functional analysis of Waardenburg syndrome-associated PAX3 and SOX10 mutations: report of a dominant-negative SOX10 mutation in Waardenburg syndrome type II.Hum Genet, 2012, 131(3): 491–503.
[57]Zhang H, Luo HJ, Chen HS, Mei LY, He CF, Jiang L, Li JD, Feng Y.Functional analysis of MITF gene mutations associated with Waardenburg syndrome type 2.FEBS Lett, 2012, 586(23): 4126–4131.
[58]Sommer L.Generation of melanocytes from neural crest cells.Pigment Cell Melanoma Res, 2011, 24(3):411–421.
[59]Lang D, Epstein JA.Sox10 and Pax3 physically interact to mediate activation of a conserved c-RET enhancer.Hum Mol Genet, 2003, 12(8): 937–945.
[60]Lang D, Chen F, Milewski R, Li J, Lu MM, Epstein JA.Pax3 is required for enteric ganglia formation and functions with Sox10 to modulate expression of c-ret.J Clin Invest, 2000, 106(8): 963–971.
[61]Eccles MR, He S, Ahn A, Slobbe LJ, Jeffs AR, Yoon HS,Baguley BC.MITF and PAX3 play distinct roles in melanoma cell migration; outline of a "Genetic Switch"theory involving MITF and PAX3 in proliferative and invasive phenotypes of melanoma.Front Oncol, 2013, 3:229.
[62]Wollnik B, Tukel T, Uyguner O, Ghanbari A, Kayserili H, Emiroglu M, Yuksel-Apak M.Homozygous and heterozygous inheritance of PAX3 mutations causes different types of Waardenburg syndrome.Am J Med Genet A, 2003, 122A(1): 42–45.
[63]Gad A, Laurino M, Maravilla KR, Matsushita M,Raskind WH.Sensorineural deafness, distinctive facial features, and abnormal cranial bones: a new variant of Waardenburg syndrome? Am J Med Genet A, 2008,146A(14): 1880–1885.
[64]Tremblay P, Kessel M, Gruss P.A transgenic neuroanatomical marker identifies cranial neural crest deficiencies associated with the Pax3 mutant Splotch.Dev Biol, 1995, 171(2): 317–329.
[65]Wu M, Li J, Engleka KA, Zhou B, Lu MM, Plotkin JB,Epstein JA.Persistent expression of Pax3 in the neural crest causes cleft palate and defective osteogenesis in mice.J Clin Invest, 2008, 118(6): 2076–2087.
[66]Kubic JD, Young KP, Plummer RS, Ludvik AE, Lang D.Pigmentation PAX-ways: the role of Pax3 in melanogenesis, melanocyte stem cell maintenance, and disease.Pigment Cell Melanoma Res, 2008, 21(6): 627–645.
[67]Fenby BT, Fotaki V, Mason JO.Pax3 regulates Wnt1 expression via a conserved binding site in the 5' proximal promoter.Biochim Biophys Acta, 2008, 1779(2):115–121.
[68]Mayanil CS, Pool A, Nakazaki H, Reddy AC, Mania-Farnell B, Yun B, George D, McLone DG, Bremer EG.Regulation of murine TGFbeta2 by Pax3 during early embryonic development.J Biol Chem, 2006,281(34): 24544–24552.
[69]Puppo F, Griseri P, Fanelli M, Schena F, Romeo G,Pelicci P, Ceccherini I, Ravazzolo R, Patrone G.Cellline specific chromatin acetylation at the Sox10-Pax3 enhancer site modulates the RET proto-oncogene expression.FEBS Lett, 2002, 523(1–3): 123–127.
[70]Conway SJ, Bundy J, Chen J, Dickman E, Rogers R,Will BM.Decreased neural crest stem cell expansion is responsible for the conotruncal heart defects within the splotch (Sp(2H))/Pax3 mouse mutant.Cardiovasc Res,2000, 47(2): 314–328.
[71]Morgan SC, Relaix F, Sandell LL, Loeken MR.Oxidative stress during diabetic pregnancy disrupts cardiac neural crest migration and causes outflow tract defects.Birth Defects Res A Clin Mol Teratol, 2008, 82(6):453–463.
[72]Soma T, Tsuji Y, Hibino T.Involvement of transforming growth factor-beta2 in catagen induction during the human hair cycle.J Invest Dermatol, 2002, 118(6):993–997.
[73]Taneyhill LA, Bronner-Fraser M.Dynamic alterations in gene expression after Wnt-mediated induction of avian neural crest.Mol Biol Cell, 2005, 16(11): 5283– 5293.
[74]Schreiner S, Cossais F, Fischer K, Scholz S, Bösl MR,Holtmann B, Sendtner M, Wegner M.Hypomorphic Sox10 alleles reveal novel protein functions and unravel developmental differences in glial lineages.Development, 2007, 134(18): 3271–3281.
[75]Wissmüller S, Kosian T, Wolf M, Finzsch M, Wegner M.The high-mobility-group domain of Sox proteins interacts with DNA-binding domains of many transcription factors.Nucleic Acids Res, 2006, 34(6): 1735–1744.
[76]Pingault V, Guiochon-Mantel A, Bondurand N, Faure C,Lacroix C, Lyonnet S, Goossens M, Landrieu P.Peripheral neuropathy with hypomyelination, chronic intestinal pseudo-obstruction and deafness: a developmental"neural crest syndrome" related to a SOX10 mutation.Ann Neurol, 2000, 48(4): 671–676.
[77]Sánchez-Mejí as A, Watanabe Y, Fernández RM,López-Alonso M, Antiñolo G, Bondurand N, Borrego S.Involvement of SOX10 in the pathogenesis of Hirschsprung disease: report of a truncating mutation in an isolated patient.J Mol Med (Berl), 2010, 88(5): 507–514.
[78]Bondurand N, Kuhlbrodt K, Pingault V, Enderich J, Sajus M, Tommerup N, Warburg M, Hennekam RC, Read AP, Wegner M, Goossens M.A molecular analysis of the yemenite deaf-blind hypopigmentation syndrome:SOX10 dysfunction causes different neurocristopathies.Hum Mol Genet, 1999, 8(9): 1785–1789.
[79]Harris ML, Baxter LL, Loftus SK, Pavan WJ.Sox proteins in melanocyte development and melanoma.Pigment Cell Melanoma Res, 2010, 23(4): 496–513.
[80]Wahlbuhl M, Reiprich S, Vogl MR, Bösl MR, Wegner M.Transcription factor Sox10 orchestrates activity of a neural crest-specific enhancer in the vicinity of its gene.Nucleic Acids Res, 2012, 40(1): 88–101.
[81]Southard-Smith EM, Kos L, Pavan WJ.Sox10 mutation disrupts neural crest development in Dom Hirschsprung mouse model.Nat Genet, 1998, 18(1): 60–64.
[82]Kuhlbrodt K, Herbarth B, Sock E, Hermans-Borgmeyer I, Wegner M.Sox10, a novel transcriptional modulator in glial cells.J Neurosci, 1998, 18(1): 237–250.
[83]Breuskin I, Bodson M, Thelen N, Thiry M, Borgs L,Nguyen L, Lefebvre PP, Malgrange B.Sox10 promotes the survival of cochlear progenitors during the establishment of the organ of Corti.Dev Biol, 2009, 335(2):327–339.
[84]Watanabe K, Takeda K, Katori Y, Ikeda K, Oshima T,Yasumoto K, Saito H, Takasaka T, Shibahara S.Expression of the Sox10 gene during mouse inner ear development.Brain Res Mol Brain Res, 2000, 84(1–2):141–145.
[85]Pingault V, Girard M, Bondurand N, Dorkins H, Van Maldergem L, Mowat D, Shimotake T, Verma I,Baumann C, Goossens M.SOX10 mutations in chronic intestinal pseudo-obstruction suggest a complex physiopathological mechanism.Hum Genet, 2002, 111(2):198–206.
[86]Barnett CP, Mendoza-Londono R, Blaser S, Gillis J,Dupuis L, Levin AV, Chiang PW, Spector E, Reardon W.Aplasia of cochlear nerves and olfactory bulbs in association with SOX10 mutation.Am J Med Genet A, 2009,149A(3): 431–436.
[87]Mao YY, Reiprich S, Wegner M, Fritzsch B.Targeted deletion of Sox10 by Wnt1-cre defects neuronal migration and projection in the mouse inner ear.PLoS ONE,2014, 9(4): e94580.
[88]Otreba M, Miliński M, Buszman E, Wrześniok D, Beberok A.Hereditary hypomelanocytoses: the role of PAX3, SOX10, MITF, SNAI2, KIT, EDN3 and EDNRB genes.Postepy Hig Med Dosw(Online), 2013, 67:1109–1118.
[89]Betters E, Liu Y, Kjaeldgaard A, Sundstrom E, García-Castro MI.Analysis of early human neural crest development.Dev Biol, 2010, 344(2): 578–592.
[90]Cheli Y, Ohanna M, Ballotti R, Bertolotto C.Fifteen-year quest for microphthalmia-associated transcription factor target genes.Pigment Cell Melanoma Res,2010, 23(1): 27–40.
[91]Hou L, Pavan WJ.Transcriptional and signaling regulation in neural crest stem cell-derived melanocyte development: do all roads lead to Mitf? Cell Res, 2008,18(12): 1163–1176.
[92]Hou L, Panthier JJ, Arnheiter H.Signaling and transcriptional regulation in the neural crest-derived melanocyte lineage: interactions between KIT and MITF.Development, 2000, 127(24): 5379–5389.
[93]Wan P, Hu Y, He L.Regulation of melanocyte pivotal transcription factor MITF by some other transcription factors.Mol Cell Biochem, 2011, 354(1–2): 241–246.
[94]Bondurand N, Pingault V, Goerich DE, Lemort N, Sock E, Le Caignec C, Wegner M, Goossens M.Interaction among SOX10, PAX3 and MITF, three genes altered in Waardenburg syndrome.Hum Mol Genet, 2000, 9(13):1907–1917.
[95]Ludwig A, Rehberg S, Wegner M.Melanocyte-specific expression of dopachrome tautomerase is dependent on synergistic gene activation by the Sox10 and Mitf transcription factors.FEBS Lett, 2004, 556(1–3): 236–244.
[96]Hou L, Arnheiter H, Pavan WJ.Interspecies difference in the regulation of melanocyte development by SOX10 and MITF.Proc Natl Acad Sci USA, 2006, 103(24):9081–9085.
[97]Zhu L, Lee HO, Jordan CS, Cantrell VA, Southard-Smith EM, Shin MK.Spatiotemporal regulation of endothelin receptor-B by SOX10 in neural crest-derived enteric neuron precursors.Nat Genet, 2004, 36(7): 732–737.
[98]Sato-Jin K, Nishimura EK, Akasaka E, Huber W, Nakano H, Miller A, Du J, Wu M, Hanada K, Sawamura D,Fisher DE, Imokawa G.Epistatic connections between microphthalmia-associated transcription factor and endothelin signaling in Waardenburg syndrome and other pigmentary disorders.FASEB J, 2008, 22(4): 1155–1168.
[99]Sarkozy A, Digilio MC, Dallapiccola B.Leopard syndrome.Orphanet J Rare Dis, 2008, 3: 13.
[100]Schaeper U, Gehring NH, Fuchs KP, Sachs M, Kempkes B, Birchmeier W.Coupling of Gab1 to c-Met, Grb2, and Shp2 mediates biological responses.J Cell Biol, 2000,149(7): 1419–1432.
[101]Mascarenhas JB, Littlejohn EL, Wolsky RJ, Young KP,Nelson M, Salgia R, Lang D.PAX3 and SOX10 activate MET receptor expression in melanoma.Pigment Cell Melanoma Res, 2010, 23(2): 225–237.
[102]Beuret L, Flori E, Denoyelle C, Bille K, Busca R,Picardo M, Bertolotto C, Ballotti R.Up-regulation of MET expression by α-melanocyte-stimulating hormone and MITF allows hepatocyte growth factor to protect melanocytes and melanoma cells from apoptosis.J Biol Chem, 2007, 282(19): 14140–14147.
[103]Sarkozy A, Carta C, Moretti S, Zampino G, Digilio MC,Pantaleoni F, Scioletti AP, Esposito G, Cordeddu V, Lepri F, Petrangeli V, Dentici ML, Mancini GM, Selicorni A, Rossi C, Mazzanti L, Marino B, Ferrero GB, Silengo MC, Memo L, Stanzial F, Faravelli F, Stuppia L,Puxeddu E, Gelb BD, Dallapiccola B, Tartaglia M.Germline BRAF mutations in Noonan, LEOPARD,and cardiofaciocutaneous syndromes: molecular diversity and associated phenotypic spectrum.Hum Mutat,2009, 30(4): 695–702.
[104]Fraser FC, Sproule JR, Halal F.Frequency of the branchio-oto-renal (BOR) syndrome in children with profound hearing loss.Am J Med Genet, 1980, 7(3):341–349.
[105]Martínez-Frías ML, Bermejo Sánchez E, Arroyo Carrera I, Pérez Fernández JL, Pardo Romero M, Burón Martínez E, Hernández Ramón F.The Townes-Brocks syndrome in Spain: the epidemiological aspects in a consecutive series of cases.An Esp Pediatr,1999, 50(1): 57–60.
[106]Chang EH, Menezes M, Meyer NC, Cucci RA, Vervoort VS, Schwartz CE, Smith RJH.Branchio-oto-renal syndrome: the mutation spectrum in EYA1 and its phenotypic consequences.Hum Mutat, 2004, 23(6): 582–589.
[107]Kohlhase J, Wischermann A, Reichenbach H, Froster U,Engel W.Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome.Nat Genet,1998, 18(1): 81–83.
[108]Wong EY, Ahmed M, Xu PX.EYA1-SIX1 complex in neurosensory cell fate induction in the mammalian inner ear.Hear Res, 2013, 297: 13–19.
[109]Brodbeck S, Englert C.Genetic determination of nephrogenesis: the Pax/Eya/Six gene network.Pediatr Nephrol, 2004, 19(3): 249–255.
[110]Alasti F, Sadeghi A, Sanati MH, Farhadi M, Stollar E,Somers T, Van Camp G.A mutation in HOXA2 is responsible for autosomal-recessive microtia in an Iranian family.Am J Hum Genet, 2008, 82(4): 982–991.
[111]Hauptmann G, Belting HG, Wolke U, Lunde K, Söll I,Abdelilah-Seyfried S, Prince V, Driever W.Spiel ohne grenzen/pou2 is required for zebrafish hindbrain segmentation.Development, 2002, 129(7): 1645–1655.
[112]Massa V, Gaudenzi G, Sangiorgio L, Cotelli F, Giavini E.Krox20 is down-regulated following triazole in vitro embryonic exposure: a polycompetitor-based assay.Toxicol Lett, 2007, 169(3): 196–204.
[113]Reiprich S, Kriesch J, Schreiner S, Wegner M.Activation of Krox20 gene expression by Sox10 in myelinating Schwann cells.J Neurochem, 2010, 112(3): 744–754.
[114]Marín F, Charnay P.Hindbrain patterning: FGFs regulate Krox20 and mafB/kr expression in the otic/preotic region.Development, 2000, 127(22): 4925–4935.
[115]Weisinger K, Kayam G, Missulawin-Drillman T,Sela-Donenfeld D.Analysis of expression and function of FGF-MAPK signaling components in the hindbrain reveals a central role for FGF3 in the regulation of Krox20, mediated by Pea3.Dev Biol, 2010, 344(2):881–895.
[116]Madden C, Halsted MJ, Hopkin RJ, Choo DI, Benton C,Greinwald JH Jr.Temporal bone abnormalities associated with hearing loss in Waardenburg syndrome.Laryngoscope, 2003, 113(11): 2035–2041.
[117]Elmaleh-Bergès M, Baumann C, Noël-Pétroff N, Sekkal A, Couloigner V, Devriendt K, Wilson M, Marlin S,Sebag G, Pingault V.Spectrum of temporal bone abnormalities in patients with Waardenburg syndrome and SOX10 mutations.AJNR Am J Neuroradiol, 2013, 34(6):1257–1263.
[118]Stinckens C, Standaert L, Casselman JW, Huygen PL,Kumar S, Van de Wallen J, Cremers CW.The presence of a widened vestibular aqueduct and progressive sensorineural hearing loss in the branchio-oto-renal syndrome.A family study.Int J Pediatr Otorhinolaryngol,2001, 59(3): 163–172.
[119]Kemperman MH, Stinckens C, Kumar S, Huygen PL,Joosten FB, Cremers CW.Progressive fluctuant hearing loss, enlarged vestibular aqueduct, and cochlear hypoplasia in branchio-oto-renal syndrome.Otol Neurotol,2001, 22(5): 637–643.
[120]Propst EJ, Blaser S, Gordon KA, Harrison RV, Papsin BC.Temporal bone findings on computed tomography imaging in branchio-oto-renal syndrome.Laryngoscope,2005, 115(10): 1855–1862.
[121]Ito T, Noguchi Y, Yashima T, Kitamura K.SIX1 mutation associated with enlargement of the vestibular aqueduct in a patient with branchio-oto syndrome.Laryngoscope, 2006, 116(5): 796–799.
