新型有序介孔炭基固体酸的制备及其催化双酚A合成
2014-04-09山文斌董秀芹张敏华
山文斌,董秀芹,张敏华*
(1.天津大学石油化工技术开发中心,天津 300072;2.天津大学绿色合成与转化重点实验室,天津300072)
双酚A(Bisphenol A,BPA)是重要的有机化工原料,主要用于生产环氧树脂、聚碳酸酯等多种高分子材料[1]。由苯酚和丙酮在酸催化剂作用下经缩合反应制得。

工业上双酚A生产主要有硫酸法、氯化氢法和离子交换树脂法。硫酸法、氯化氢法因副产物多,污染严重,后处理复杂,设备腐蚀严重,已基本淘汰;目前广泛应用的是离子交换树脂法。据报道,巯基改性离子交换树脂(如Amberlyst)在缩合反应中显示出优异的催化活性,丙酮转化率高达90~100%,双酚A选择性接近90%[2-3]。但热稳定性较差、易被副产物水污染中毒、易溶胀破裂等缺点严重影响树脂催化剂的寿命[4]。另外,杂多酸[5]、新型固体酸[6]、分子筛[7]、功能化介孔硅[8]、离子液体[9]等催化剂也已见报道,但仍处于实验室研究阶段。
炭材料以其优良的化学惰性、热稳定性、疏水性受到广泛关注。Hara等[10-12]以萘等多环芳烃[10]、葡萄糖、蔗糖[11]、微晶纤维素[13-16]、木屑[17]为碳源,经炭化、磺化制备出高酸密度、高效稳定的炭基固体酸。但该催化剂比表面积过低(2 m2/g),不适于大分子、疏水型反应物参与的反应。因此,介孔炭基固体酸因其孔径可调变、表面疏水性、易功能化等优点,成为大分子反应进行酸催化反应的有效选择。
近年来介孔炭基固体酸的报道甚多[18-21],但未将其用于双酚A合成反应中。本研究以模板法制备介孔炭基固体酸,系统探讨了不同制备条件对所制得介孔炭基固体酸催化剂比表面积、孔道结构、表面酸性的影响,并用于双酚A合成反应中,探讨不同制备方法对催化剂催化活性的影响,以期制备出新型高效、疏水稳定、高度有序的介孔炭基固体酸催化剂。
1 实验部分
1.1 试剂与原料
乙醇、甲醛、苯酚、氢氧化钠、氯化钠、盐酸:分析纯,天津市光复精细化工研究所;丙酮、浓硫酸(质量分数98%):天津市江天化工技术有限公司;F127(MW=13 388,PEO106PPO70PEO106):Sigma-Aldrich公司。
1.2 催化剂的制备
模板法介孔炭的制备[22]:将0.13 g NaOH溶液(质量分数为 20%,0.65 mmol)加入到 0.61 g(6.5 mmol)苯酚和1.05 g甲醛溶液(质量分数为37%,13.0 mmol)混合物中,70 ℃下搅拌 1 h,得到前驱体溶液。将1 g模板剂F127溶解于20 g无水乙醇中,然后加入前驱体溶液搅拌10 min,转至圆盘内在室温下放置5~8 h,100℃热聚合24 h。热聚合结束后,N2气氛下以1℃/min升至350℃,煅烧5 h,脱除模板剂 F127,形成具有多孔结构的介孔炭C-FDU-15。
介孔炭基固体酸的制备:取一定量介孔炭置于管式炉中,N2(40 mL/min)保护下,以 2℃/min升至不同温度(400~700℃)炭化3 h,得到黑色固体,记作FDU-15-TC(TC表示炭化温度)。将炭化后样品放入聚四氟内衬的水热釜中,加入浓硫酸,以2℃/min升至180℃磺化10 h。自然冷却至室温,经过滤、洗涤,60℃下干燥8 h后,即可得到介孔炭基固体酸FDU-15-TC-S(S表示磺化后)。
1.3 催化剂的表征
采用美国Micromeritics公司生产的Tristar 3000型吸附仪测定比表面积、孔体积和孔径;采用荷兰PANalytical公司的X’PertPRO型X射线衍射仪进行XRD分析,Cu靶,小角衍射扫描范围0.5°~5.0°、扫描速率 0.5(°)/min,广角衍射扫描范围 5°~80°、扫描速率 5(°)/min,管电流30 A,电压 40 kV;采用透射电子显微镜(HRTEM,FEITecnai G2F20)观察催化剂有序介孔孔道结构;采用EDS能谱分析催化剂元素S含量;采用美国尼高力公司的Nicolet560 FT-IR分析仪表征催化剂表面官能团;采用酸碱滴定方法测定催化剂强酸量(-SO3H)和总酸量。
1.4 催化剂的评价
在50 mL三口烧瓶中装入0.05 mol苯酚、0.005 mol丙酮和0.10 g催化剂,置于85℃水浴中开始反应(冷凝回流,转速为450 r/min)。反应8 h后停止搅拌,过滤催化剂。滤液采用液相色谱-质谱联用仪(Agilent 1100)进行分析。液相色谱条件:色谱柱 Zorbox C18柱(250 mm×0.46 mm,0.5 μm);柱温 20℃;流动相甲醇-水(63∶37,体积比),流速0.5 mL/min;进样量20μL;检测器 UV 278 nm。采用外标法进行定量分析。
2 结果与讨论
2.1 催化剂的表征
图1为介孔炭磺化前后的N2吸附-脱附等温线及BJH孔分布曲线。可以看出,炭化后,不同的炭化温度样品均具有典型的Ⅳ型曲线和H1型滞后环,且在相对压力p/p0=0.4~0.8之间出现明显的毛细凝聚现象,说明材料具有均一的介孔结构。由BJH孔分布曲线可以看出在~4 nm左右有1个很窄的孔径分布,进一步说明介孔孔道结构的有序性。且随着炭化温度的升高,孔径分布越窄,介孔孔道有序性越高。但磺化后,不同炭化温度样品均发生了不同程度的坍塌(表1)。特别是样品FDU-15-400-S[图1a)],磺化后介孔孔道几乎完全坍塌,BET比表面由炭化前的525.0 m2/g降到22.6 m2/g。这可能是因为部分炭化情况下,炭骨架氧氢含量太高,浓硫酸剧烈的磺化过程造成炭骨架的破坏,孔道坍塌。而随着炭化温度升高,炭化程度逐渐增强,骨架内氧氢含量相应降低,磺化过程对介孔孔道造成的影响明显减弱[图1c)和1d)]。

图1 介孔炭磺化前后的N2吸附-脱附等温线Fig.1 N2 adsorp tion-desorption isotherms of mesoporous carbon before and after su lfonation

表1 介孔炭磺化前后的体相性质Tab le 1 Textural p roperties of mesoporous carbon before and after su lfonation
图2是不同炭化温度下介孔炭磺化前后的XRD小角衍射图。不同炭化温度下介孔炭磺化前后样品(10)峰对应的d10值以及通过公式a0=2d10/计算得到的对应结构单元参数a0见表1。不同炭化温度下样品均显示出3个衍射峰,记作(10)、(20)和(21)峰,表明介孔炭磺化前后均属于二维六方(p6mm)晶系,且具有高度有序的介孔结构。磺化后[图2b)],样品仍具有高度有序的介孔结构。但随着炭化温度的升高,(10)峰的峰强度逐渐增强,介孔孔道有序性也逐渐增强[23]。说明磺化过程对较低炭化温度样品的介孔孔道有序性影响较大,易造成孔道坍塌;而对于较高炭化温度的样品,影响相对较小,仍保持高度有序的介孔结构。

图2 XRD谱Fig.2 XRD patterns of mesoporous carbon a)and su lfonated mesoporous carbon b)
图3是FDU-15-500和FDU-15-700磺化前后的XRD衍射图。
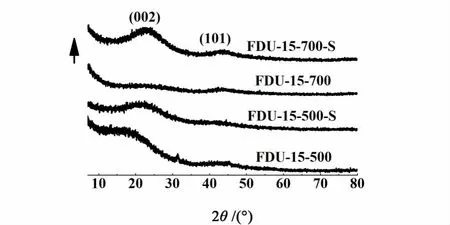
图3 介孔炭磺化前后的XRD谱Fig.3 XRD patterns of mesoporous carbon before and after su lfonation
由图3可以看出,样品的结晶度较低[24],磺化前后样品均具有(002)、(101)2个衍射峰。其中,位于2θ=10°~30°处的强衍射峰的出现,表明介孔炭骨架是由无规则的多环芳香碳薄层组成的[25-27];而2θ=40°~50°处的弱衍射峰则说明了石墨化结构的出现,而且随着炭化温度的升高,该峰强度逐渐增强[12,28]。
图4为样品FDU-15-500和 FDU-15-700磺化前后的TEM图。

图4 介孔炭磺化前后的TEM图Fig.4 TEM images of mesoporous carbon before and after su lfonation
由图4可以看出,样品 FDU-15-500、FDU-15-700磺化前后均呈现出高度有序的介孔结构。由于浓硫酸苛刻的磺化条件,低温炭化样品磺化后孔道较磺化前有明显的坍塌[图3a)和图3b)];但随着炭化温度升高,坍塌程度逐渐降低[图3c)和图3d)]。由TEM照片可以计算出4种样品相邻孔壁的间距(对应二维六角材料的 d10值)分别为10.8、10.6、10.1 和9.8 nm,这与XRD得到的结果是一致的。
图5为样品FDU-15-500-S和FDU-15-700-S的EDX能谱,通过EDX能谱对磺化后样品中的元素分析。
由图5可以看出,不同炭化温度下磺化后样品均含有C、O和S的3种元素,但高温炭化样品O和S含量明显低于低温炭化样品,说明随着炭化温度的升高,-SO3H含量逐渐降低。样品FDU-15-500-S和FDU-15-700-S中S元素质量比分别为5.564%、1.339%,可计算出2种样品-SO3H酸密度分别是1.74和0.42 mmol/g,与酸碱中和滴定测得的催化剂-SO3H密度相符(1.71和0.53 mmol/g)。分析 2 种样品的 O/S 原子比(5.6∶1、10.8∶1),均大于-SO3H中的 O/S原子比(3∶1),表明炭骨架内仍残留有环氧基、羟基和羰基等含氧基团,介孔炭基固体酸只是部分炭化,并未完全炭化[29]。

图5 介孔炭磺酸催化剂的EDX能谱Fig.5 EDX spectra of su lfonated mesoporous carbon
图6是介孔炭基固体酸催化剂的FT-IR谱。

图6 介孔炭基固体磺酸催化剂的FT-IR谱图Fig.6 FT-IR spectra of su lfonated mesoporous carbon
由图6可见,1 209 cm-1处 Ar-OH伸缩振动峰、1 466 和 1 610 cm-1处CC伸缩振动峰的存在,证明炭化后样品存在大量的多环芳香碳。但随着炭化温度的升高,Ar-OH伸缩振动峰、CC伸缩振动峰均减弱,表明高温炭化不利于多环芳香碳的形成。而1 079 cm-1处伸缩振动峰、1 220 cm-1处伸缩振动峰表明磺化后催化剂上-SO3H的存在。而1 750 cm-1处CO伸缩振动可能代表-COOH的存在。随着炭化温度的升高,炭化程度越高,代表的伸缩振动特征峰逐渐减弱,表明高温炭化不利于介孔炭基固体酸的磺化过程,不利于介孔炭基固体酸的形成。
表2列出了不同炭化温度介孔炭基固体酸的酸密度。随着炭化温度升高,-SO3H密度、总酸密度均逐渐降低。由表2可知,400℃炭化,-SO3H密度、总酸密度最高;700℃炭化,-SO3H密度、总酸密度均为最低。结合 N2吸附-脱附表征结果,虽然400℃炭化时酸密度最高,但炭骨架内氧氢含量过高,磺化过程易造成介孔孔道坍塌。而700℃炭化有利于介孔孔道的保持,不易坍塌,但介孔炭骨架表面含氧基团含量较少,石墨化程度过高,不利于介孔炭的磺化,酸密度较低;而500℃炭化样品酸密度与400℃接近,分别为1.71 mmol/g、3.28 mmol/g,且在磺化后仍保持高度有序的介孔孔道结构,孔径分布较窄,孔径均一(~3.7 nm)。

表2 催化剂催化双酚A合成的催化活性Tab le 2 Catalytic activities of catalysts tested for synthesis of bisphenol A
2.2 催化剂催化双酚A合成
为了考察介孔炭基固体酸催化剂的催化活性,探究不同炭化温度对催化活性的影响,采用介孔炭基固体酸催化剂催化丙酮和苯酚缩合生成双酚A的反应。并与无定型炭基固体酸、市售的强酸型树脂001×7和强酸型大孔树脂D072进行对比,几种催化剂的活性中心均是-SO3H。
由表2可以看出,制备出的新型介孔炭基固体酸催化剂的催化活性明显高于无定型炭基固体酸和市售的强酸型离子交换树脂。其中,当强酸型树脂001×7催化缩合反应时,虽然-SO3H酸密度相当高,但催化活性几乎为0。可能由于该凝胶型树脂孔道较小,属于显微孔,反应物只能与催化剂表面的少量活性位进行反应,无法接触到催化剂内部绝大部分活性位,催化活性相当低[30]。而强酸型大孔树脂D072虽然满足了较高的酸密度和大孔孔道结构两个条件,但催化活性仍然很低,可能是由于树脂疏水性较差,活性中心易与缩合反应中生成的副产物水发生水和作用而失活[3,5]。相比之下,炭基固体酸催化剂由于其炭基质的原因,疏水性较强酸型树脂有明显提高。但是无定型炭基固体酸催化剂由于其比表面积过低(<2 m2/g),几乎无孔道,限制了其在大分子疏水型反应中的应用,催化活性相当低。
对比几种介孔炭基固体酸催化剂发现,介孔炭基固体酸催化剂的催化活性与比表面积与酸密度相关,较高的酸密度和比表面积,有利于反应物分子进入有序介孔孔道接触到介孔炭基固体酸表面的活性位-SO3H。其中,FDU-15-400-S由于磺化后大部分孔道已经坍塌,比表面积过低,虽然酸密度最高,但催化活性不高。而 FDU-15-500-S、FDU-15-600-S、FDU-15-700-S三种样品比表面积相差不大,但随着炭化温度的升高,-SO3H酸密度急剧降低,催化活性同样逐渐降低。因此,当炭化温度为500℃时,介孔炭基固体酸酸密度较高,且磺化后介孔孔道仍然高度有序,催化活性最高(图7)。

图7 介孔炭基固体酸催化双酚A合成催化活性图Fig.7 Catalytic activity of sulfonated mesoporous carbon as a function of carbonization temperatu re for bisphenol A synthesis
3 结论
以模板法制备高度有序的介孔炭基固体酸催化剂,考察了不同炭化温度对催化剂介孔结构、表面酸性以及催化活性的的影响,从而制备出具有有序介孔孔道、高密度-SO3H、高催化活性的固体酸催化剂。结果表明:炭化温度的选择对介孔炭基固体酸催化剂的介孔孔道结构及表面酸性具有重要的影响。高温炭化有利于在磺化过程中介孔孔道的保持,不易坍塌,但石墨化程度过高,不利于介孔炭的磺化,酸密度较低;而低温炭化有利于介孔炭的磺化,酸密度高,但苛刻的磺化过程易造成介孔孔道的大范围坍塌。而500℃炭化样品磺化后仍保持高度有序的介孔孔道结构,比表面积较大(393 m2/g),酸密度较高(1.71 mmol/g)。
较强的酸性、较大的比表面积、高度有序的介孔孔道以及疏水的骨架结构使得新型有序介孔炭基固体酸催化剂在催化苯酚与丙酮缩合生成双酚A的反应中具有明显的优势,催化活性高于无定型炭基固体酸和市售的大孔强酸型树脂,使其作为固体酸催化剂在双酚A领域具有较好的应用前景。
参考文献:
[1] Yadav G D,Salgaonkar S S.Loss prevention and waste minimization with cascade-engineered green synthesis of bisphenol-A from cumene hydroperoxide and phenol using heteropoly acid-supported clay catalysts[J].Organic Process Research&Development,2009,13: 501-509
[2] Angelis A,Inallina P,Perego C.Synthesis of bisphenol-A from phenol&acetone[J].Ind Eng Res Dev,2004,43:1 169-1 178
[3] Chen C,Cheng S,Jang L.Dual-Functionalized large poremesoporous silica as an efficient catalyst for biephenol-A synthesis[J].Microporous and Mesoporous Materials.2008,109:258-270
[4] 马怡,常春,李洪亮.合成双酚A催化剂研究新进展[J].化工进展.2007,12(26):1 686-1 689 Ma Yi,Chang Chun,Li Hongliang.New progress on research of catalysts for synthesis of bisphenol A[J].Chemical Industry and Engineering Progress,2007,12(26): 1 686-1 689(in Chinese)
[5] Shimizu K,Kontani S,Yamada S,et al.Design of active centers for bisphenol-A synthesis by organic-inorganic dual modification of heteropolyacid[J].Applied Catalysis A: General,2010,380:33-39
[6] Hou L,Cai Q,Lu B,et al.A novel solid acid for synthesis of bisphenol A[J].Catalysis Letters,2006,111:153-157
[7] Nowinska K,Kaleta W.Synthesis of bisphenol-A over heteropoly compounds encapsulated intomesoporousmolecular sieves[J].Applied Catalysis A: General,2000,203:91-100
[8] Margelefsky E L,Zeidan R K,Dufaud V,et al.Organized surface functional groups:Cooperative catalysis via thiol/sulfonic acid pairing[J].J Am Chem Soc,2007,129:13 691-13 697
[9] Jia L,Hua C,Dai L,et al.Synthesis of bisphenol A catalyzed by Et3NHCl-AlCl3ionic liquids[J].React Kinet Catal Lett,2004,2(81): 235-240
[10] Hara M,Yoshida T,Takagaki A,et al.A carbon material as a strong protonic acid[J].Angew Chemint Ed,2004,43,2 955-2 958
[11] Toda M,Takagaki A,Okamura M,et al.Biodieselmade with sugar catalyst[J].Nature,2005,438: 178-179
[12] Okamura M,Takagaki A,Toda M,et al.Acid-Catalyzed reactions on flexible polycyclic aromatic carbon in amorphous carbon[J].Chem Mater,2006,18,3 039-3 045
[13] Fukuhara K,Nakajima K,Kitano M,et al.Structure and catalysis of cellulose-derived amorphous carbon bearing SO3H groups[J].Chem Sus Chem,2011,4:778-784
[14] Suganuma S,Nakajima K,Kitano M,et al.Hydrolysis of cellulose by amorphous carbon bearing SO3H,COOH,and OH groups[J].J Am Chem Soc,2008,130:12 787-12 793
[15] Kitano M,Yamaguchi D,Suganuma S,et al.Adsorption-Enhanced hydrolysis ofβ-1,4-glucan on graphenebased amorphous carbon bearing SO3H,COOH,and OH groups[J].Langmuir,2009,25(9): 5 068-5 075
[16] Yamaguchi D,Kitano M,Suganuma S,et al.Hydrolysis of cellulose by a solid acid catalyst under optimal reaction conditions[J].J Phys Chem C,2009,113:3 181-3 188
[17] Kitano M,Arai K,Kodama A,et al.Preparation of a sulfonated porous carbon catalystwith high specific surface area[J].Catalysis Letters,2009,131(1): 242-249
[18] Geng L,Yu G,Wang Y,et al.Ph-SO3H-modified mesoporous carbon as an efficient catalyst for the esterification of oleic acid[J].Applied Catalysis A: General,2012,427/428:137-144
[19] Zareyee D,GhandaliM S,Khalilzadeh M A.Sulfonated ordered nanoporous carbon(CMK-5-SO3H)as an efficient and highly recyclable catalyst for the silylation of alcohols and phenols with hexamethyldisilazane(HMDS)[J].Catal Lett,2011,141: 1 521-1 525
[20] Suganuma S,Nakajima K,Kitano M,et al.SO3H-bearing mesoporous carbon with highly selective catalysis[J].M icroporous and Mesoporous Materials,2011,143:443-450
[21] Mayes R T,Fulvio R F,Ma Z,et al.Phosphorylated mesoporous carbon as a solid acid catalyst[J].Phys Chem Chem Phys,2011,13: 2 492-2 494
[22] Meng Y,Gu D,Zhang F,et al.Ordered mesoporous polymers and homologous carbon frameworks:Amphiphilic surfactant temp lating and direct transformation[J].Angew Chemint Ed,2005,44:7 053-7 059
[23] Xing R,Liu Y,Wang Y,et al.Active solid acid catalysts prepared by sulfonation of carbonization-controlled mesoporous carbon materials[J].Microporous and Mesoporous Materials,2007: 105: 41-48
[24] Geng L,Wang Y,Yu G,et al.Efficient carbon-based solid acid catalysts for the esterification of oleic acid[J].Catalysis Communications,2011,13(1): 26-30
[25] Tsubouchi N,Xu K,Ohtsuka Y.Carbon crystallization during high-temperature pyrolysis of coals and the enhancement by calcium[J].Energy Fuels,2003,17(5): 1 119-1 125
[26] Takagaki A,Toda M,Okamura M,et al.Esterification of higher fatty acids by a novel strong solid acid[J].Catalysis Today,2006: 157-161
[27] Nakajima K,Okamura M,Kondo J N,et al.Amorphous carbon bearing sulfonic acid groups in mesoporous silica as a selective catalyst[J].Chem Mater,2009,21:186-193
[28] Liu Y,Chen J,Yao J,et al.Preparation and properties of sulfonated carbon-silica composites from sucrose dispersed on MCM-48[J].Chemical Engineering Journal,2009,148(1): 201-206
[29] Ji J,Zhang G,Chen H,et al.Sulfonated graphene as water-tolerant solid acid catalyst[J].Chemical Science,2011: 2(3): 484-487
[30] Hamdan H,Navijanti V,Nur H,et al.Fe(III)-salen encapsulated A l-MCM-41 as a catalyst in the polymerisation of bisphenol-A[J].Solid State Sciences,2005,7:239-244
