MDM2与抑制剂PDIQ 作用机制的结合自由能计算研究
2014-03-20时术华张少龙张庆刚
时术华,张少龙,张庆刚
(1.山东建筑大学理学院,济南250101;2.山东师范大学物理与电子科学学院,济南250014)
1 引 言
近期的医学临床研究表明全世界众多癌症患者的体内基本上都发现抑癌蛋白p53野生型功能的丢失.p53抑癌功能的丢失有两种公认的机制:(1)p53 蛋白内部的定点突变;(2)肿瘤蛋白MDM2通过副反馈的方式与p53相互作用,抑制了p53的功能.研究发现50%的癌症患者体内存在MDM2 的 过 度 表 达[1,2].因 此 干 扰p53 与MDM2的结合成为癌症治疗的新途径.
MDM2与p53复合物的结晶结构表明p53的三个疏水性残基Phe19,Trp23 和Leu26 插入MDM2疏水性裂缝,从而与MDM2 形成疏水性相互作用.用于治疗癌症的药物基本上都模仿了这个作用模式.几个课题组设计的肽类抑制剂能与MDM2产生较强的相互作用,拥有纳摩尔数量级的抑制效果[3,4].Phan等人设计合成的肽类抑制剂PDIQ 展现了不错的抑制效果,其IC50值达到8nM[5].该结果表明抑制剂PDIQ 对MDM2有很强的抑制效果.故阐明该抑制剂与MDM2的作用机制有助于新型高效抑制剂的研发.
分子动力学模拟和结合自由能计算已经成为研究抑制剂与蛋白质相互作用的重要工具[6-8].尽管Phan等人从结构上解释了抑制剂PDIQ 的抑制能力,但未从原子层次上阐明抑制剂PDIQ 与MDM2的作用机制.因此本工作将蛋白质库的晶格结构 (3JZS)用作动力学模拟的初始结构,且采用已成功用于研究生物体系的MM-PBSA 方法计算抑制剂PDIQ 与MDM2的结合自由能,解释二者的结合模式[9-12],从原子层次上研究了PDIQ与MDM2 的作用机制和MDM2 内部残基运动.同时采用基于残基的自由能分解方法计算抑制剂-残基相互作用以便阐明PDIQ-MDM2复合物的结构-亲和能关系.我们期望该研究能为以p53-MDM2相互作用为靶标的抗癌药物设计提供理论上的指导.
2 模型与计算
2.1 分子动力学模拟
用于动力学模拟的初始构象取自蛋白质库的晶体结构 (3JZS).图1 给出PDIQ-MDM2 复合体的结构.复合物X-射线结构中的水分子保留在初始化模型中,采用Amber 12 中的leap模块添加晶体结构中缺失的氢原子[13];复合物的力场参数由Amber 12 中 的ff03 力 场 产 生[14];PDIQMDM2复合体溶解在由5535个水分子组成的显性水合子中,水分子的模为TIP3P;两个氯离子添加到由水和复合体组成的系统中以确保整个系统维持电中性.
为消除系统中原子的不合理接触,采用Amber12中的sander模块对复合体体系执行两步的系统优化:(1)采用150kcal/ (mol·Å2)的力常数约束溶质,以便优化溶剂和中和离子;(2)无约束地优化整个系统:每一步优化均先执行2000步的最陡下降优化,接着执行4000步的共轭梯度优化.然后在200ps内把系统从0K 加热到300 K,随后进行100ps的常温300K,常压1标准大气压的动力学平衡;最后是10ns的无约束分子动力学模拟.模拟期间采用SHAKE方法限制所有含氢原子化学键的伸缩[15],模拟积分步长为2fs,PME方法用来计算长程的静电相互作用,应用周期性边界条件以消除溶剂盒子的边缘效应,非成键相互作用的截断值为10.0Å.
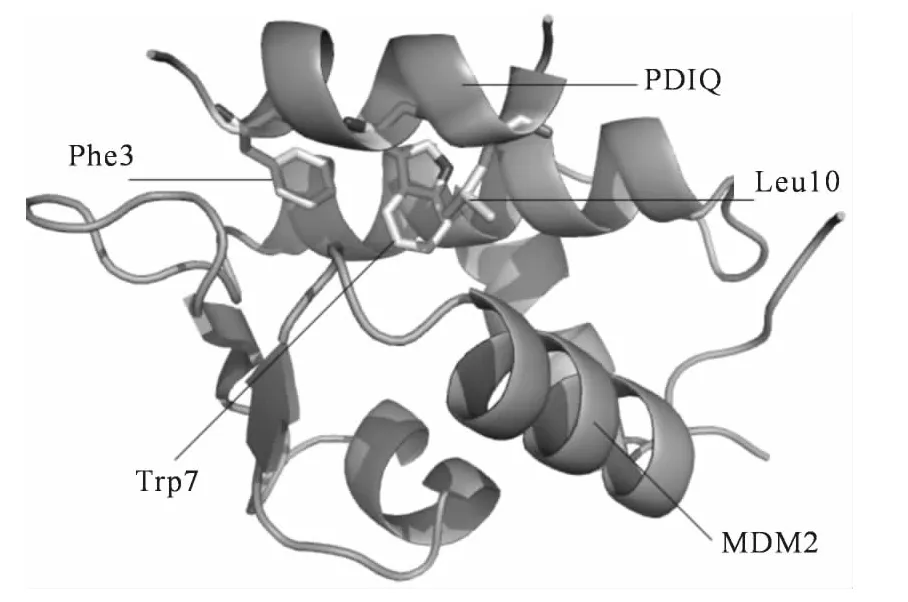
图1 PDIQ-MDM2 复合体的结构.MDM2 和PDIQ 分别用桔色和青色表示,残基Phe3′,Trp7′和Leu26′用棍棒方式显示Fig.1 Structure of PDIQ-MDM2 complex.MDM2and PDIQ are showed in orange and green,respectively,the residues Phe3′,Trp7′and Leu10′are displayed in stick mode
2.2 MM-PBSA方法计算结合自由能
采用MM-PBSA 方法计算PDIQ 与肿瘤蛋白MDM2的结合自由能.从动力学最后2ns的轨迹中每隔20ps抽取的100个构象用于结合自由能计算,结合自由能计算的公式可表示为:
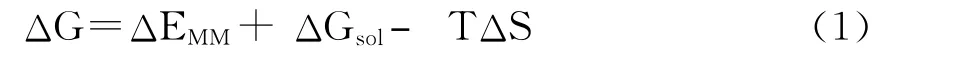
式中ΔEMM是气相中的分子力学能,ΔGsol表示溶解自由能对分子结合的贡献,TΔS表示熵变对结合自由能的贡献.ΔEMM进一步分成两部分:
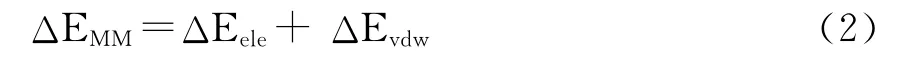
方程中的ΔEele和ΔEvdw分别表示静电相互作用和范德瓦尔斯相互作用.溶解自由能ΔGsol也分解为两个组成部分:

上式中ΔGpb和ΔGsurf分别是极性溶解自由能和非极性溶解自由能.前者可使用Amber中的pbsa方法求解泊松-玻尔兹曼方程获得,溶质和溶剂的电介常数分别设为1.0和80.0.后者由如下经验方程计算:

式中γ和β值分别取为0.00542kcal/ (mol·Å2)和0.92kcal/mol.TΔS是由于平动、转动和振动自由度变化引起的熵变对结合自由能的贡献,主要采用简振模和传统的热力学计算TΔS.

表1 MM-PBSA 计算所得到的能量 (kcal/mol)aTable 1 Energies(in kcal/mol)calculated by MM-PBSA method a
2.3 相关矩阵计算
为了研究抑制剂结合诱导的蛋白质内部运动的相关联程度,计算了相关矩阵 (Cij).这一矩阵第i行j 列的元素Cij反映了第i,j残基上Cα相对于其平均位置的涨落相关系数,通常由下列的方程确定:
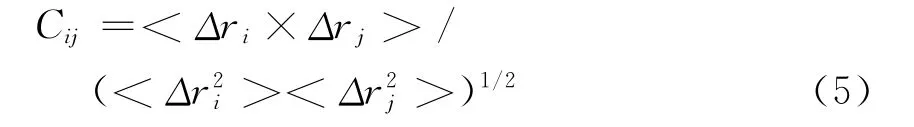
式中尖括号表示对取样的系综平均,Δri表示第i个原子相对于其平均位置的偏差,Cij的变化范围为-1到+1,正的Cij反映了残基间的相关运动,而负的Cij表述了残基间的反相关运动.
3 结果与讨论
3.1 动力学平衡的稳定性和结构的柔性
为评估分子动力学平衡的稳定性,采用Amber中的Ptraj程序计算了MDM2 主链原子相对于初始优化结构的均方根偏差 (RMSD)随时间的函数 (见图2).图2表明动力学模拟4ns后,系统达到平衡.RMSD 的平均值为1.31Å,涨落范围低于0.50Å.这表明动力学模拟平衡稳定性是可靠的.
3.2 相关运动分析
为了研究MDM2内部残基的相关运动,我们基于分子动力学模拟轨迹中原子位置,构建描述原子位置涨落的相关矩阵 (见图3).依据图3,整体而言抑制剂PDIQ 的结合并没有诱导MDM2构象的明显变化.
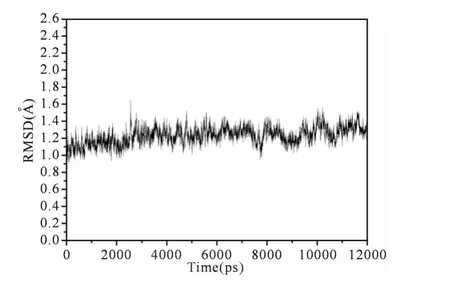
图2 MD 模拟中主链原子的均方根偏差Fig.2 Root-Mean-Square Deviation of the backbone atoms on PDIQ-MDM2 complex
从图3 可以观察到,子单元S1 和S2 内部(对角方块)发生强烈的相关运动,同时子单元S1内部也有明显的反相关运动.子单元S3 相对于S4以相关运动的趋势运动.图3表明子单元S2相对与S1和S3产生强烈的反相关运动,说明此处可能发生较大的构象变化.此外,子单元S3也与S1发生较弱的反相关运动.
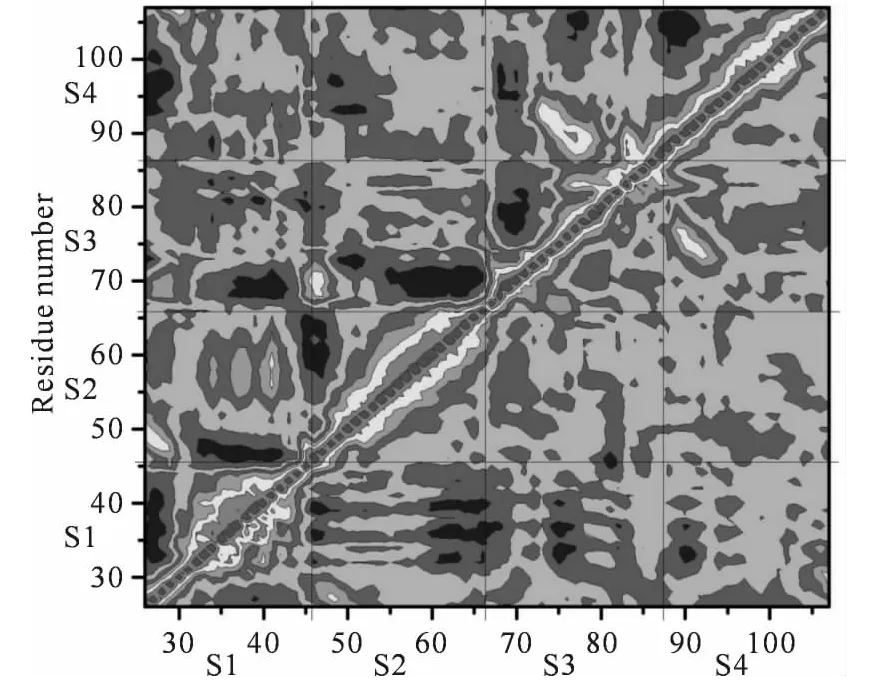
图3 系统平衡后主链Cα原子相对于其平均位置涨落的相关矩阵Fig.3 Cross-correlation matrices of the fluctuations of the coordination for Cαatoms around their mean positions after the equilibrium of system
3.3 结合自由能计算
采用MM-PBSA 方法计算了抑制剂PDIQ 与MDM2的结合自由能,其各项作用成分的贡献列在表1中.依据表1,PDIQ 与MDM2的结合自由能为-19.8kcal/mol,这表明PDIQ 与MDM2 产生较强的相互作用.
表1表明范德瓦尔斯作用能 (ΔEvdw)和非极性的溶解自由能 (ΔGsurf)分别是-67.76 和-7.80 kcal/mol.尽管气相中的静电相互作用 (ΔEele)也提供了-135.2kcal/mol的有利贡献,但是完全被不利于抑制剂结合的153.20kcal/mol的极性溶解自由能所抵消,产生了不利于抑制剂结合的极性作用 (17.99kcal/mol).由表1看到熵变对结合自由能的贡献 (-TΔS=36.78)也不利于抑制剂结合,由熵变的计算得到有利的信息,抑制剂结合前后熵明显减少,这说明抑制剂结合后结合位点原子的运动受到了很大约束.总之有利于抑制剂结合的成分中范德瓦尔斯作用占了89.7%,因此范德瓦尔斯作用驱动了PDIQ 与MDM2的结合.这个结果基本上吻合了几个课题组先前的研究[16-19].
3.4 结构-亲和能关系
为了定量地从原子层次上研究抑制剂PDIQ与MDM2的结合模式,采用基于残基的自由能分解方法计算了抑制剂-残基相互作用,相互作用谱如图4所示.依据图4,PDIQ 能与MDM2的8个残 基 Leu54,Ile61, Met62, Tyr67, Gln72,His73,Val93和His96产生强于1.4kcal/mol的相互作用,其结构亲和能关系分析如下.
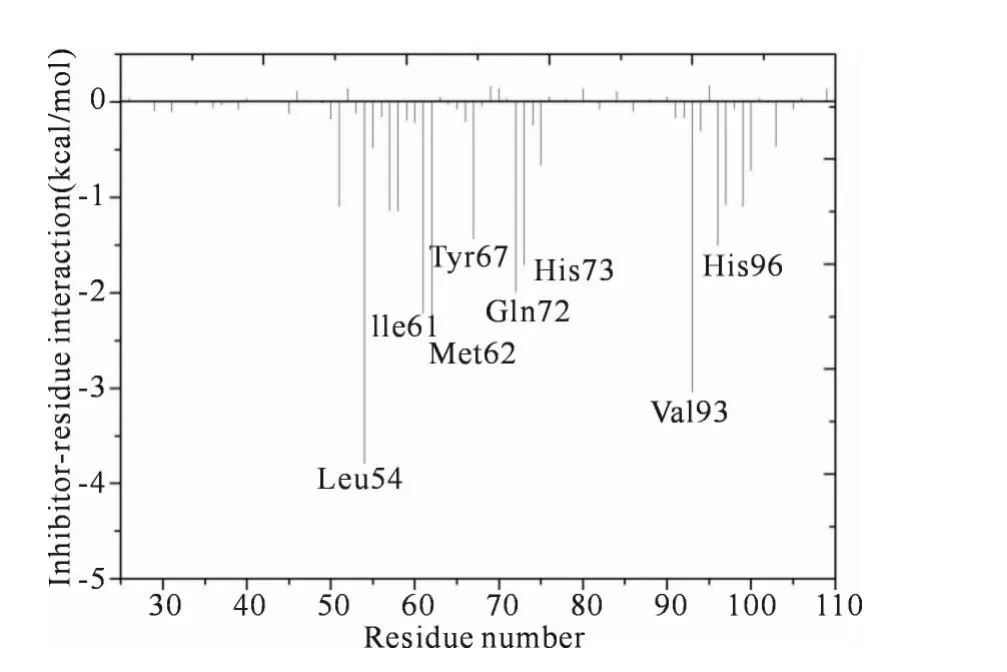
图4 抑制剂PDIQ 中关键残基与MDM2各个分离残基的相互作用谱Fig.4 Residue-residue interaction spectrum between the inhibitorp DIQ and MDM2
图4表明8个残基中Leu54与MDM2产生了最强的相互作用,其相互作用能为-3.81kcal/mol.从结构图5来看,这个较强的相互作用主要源自两个因素的贡献:(1)Trp7′疏水性环上的氮原子与残基Leu54 的羰基氧原子形成氢键 (表2);(2)Trp7′的疏水性环与Leu54 的烷基形成CH-π 相互作用.抑制剂PDIQ 与Val93的相互作用也非常强,作用能是-3.05kcal/mol.从结构上看,这个作用主要由Trp7′吲哚环与Val93的烷基间CH-π 相互作用和Leu10′的烷基与Val93烷基间的CH-CH 相互作用提供.依据图5,Met62和Ile61的烷基与残基Phe3′的苯环临近形成的CHπ相互作用分别为PDIQ 与MDM2的作用分别提供了-2.57和-2.25kcal/mol的能量贡献 (图4).基于轨迹的氢键动力学分析表明,残基Gln72与残基Phe3′形成一个氢键 (表2),这个氢键为PDIQ 与MDM2 的 相 互 作 用 提 供 了-1.99kcal/mol的贡献.残基His73与抑制剂PDIQ 的作用是-1.71kcal/mol,这与His73的5元环和Phe3′的苯环间的π-π相互作用吻合.残基Tyr67的苯环在结构上与Phe3′的苯环临近且平行,故能形成平行的π-π相互作用 (图5),该作用提供了大约-1.47kcal/mol的能量贡献.残基His96与PDIQ的作用是-1.5kcal/mol,该作用主要来自His96的疏水性5元环与Leu10′烷基间的CH-π 相互作用.上述分析结果基本上吻合几个课题组先前的工作[17-21].
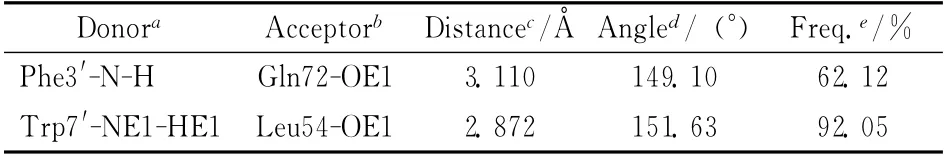
表2 主要残基的氢键分析Table 2 The hydrogen bonds of the key residues
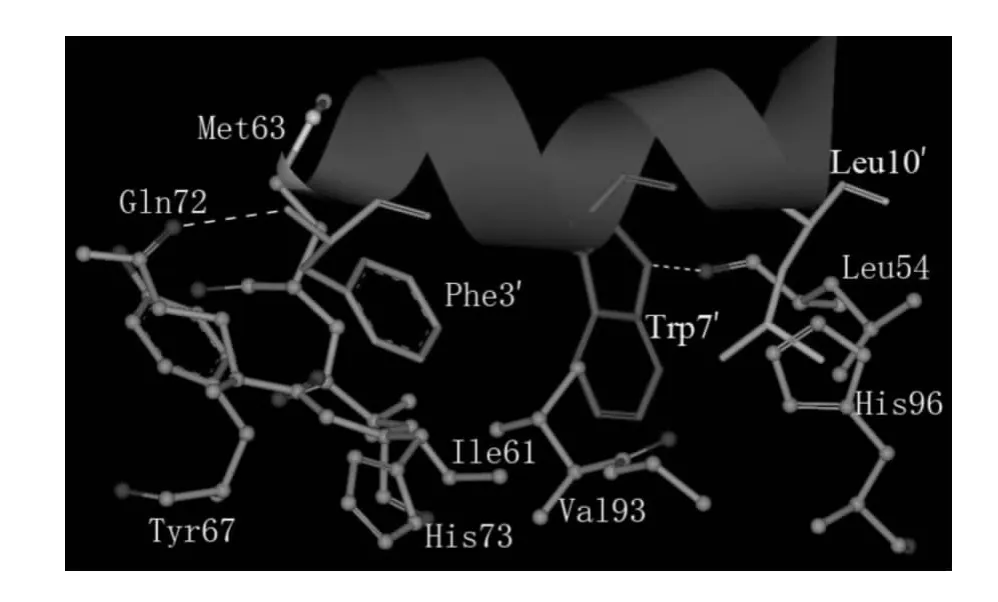
图5 pDIQ-MDM2复合体中主要残基的相对位置以及形成的氢键Fig.5 Relative geometries of the key residues in PDIQ-MDM2complex and hydrogen bonds
综合以上的结构亲和能关系可以发现,残基Ile61,Met62,Tyr67,Gln72 和His73 形成抑制剂残基Phe3′的疏水性结合口袋;残基Leu54,His73和Val93形成抑制剂残基Tyr7′的疏水性结合口袋;PDIQ 残基Leu10′的疏水性结合裂缝由残基Leu54,Val93和His96构成.在这些结合口袋中主要的结合方式为CH-CH,CH-π和π-π相互作用.因此这些作用驱动了抑制剂与MDM2的结合.
4 结 语
基于动力学轨迹的相关矩阵分析表明抑制剂结合诱导了反相关运动的主体形式.结合自由能的计算结果表明范德瓦尔斯作用主导了抑制剂与MDM2的结合.基于残基的自由能分解揭示了MDM2疏水性结合口袋内的主要作用形式,CHCH,CH-π 和π-π 相 互 作 用 驱 动 了 抑 制 剂 与MDM2的结合.这个研究结果能为抗癌药物的设计提供一定的理论启示.
[1] Chen H F,Luo R.Binding induced folding in p53-MDM2complex[J].J.Am.Chem.Soc.,2007,129:2930.
[2] Ding K,Lu Y,Nikolovska-Coleska Z,et al.Structure-based design of spiro-oxindoles as potent,specific small-molecule inhibitors of the MDM2-p53interaction[J].J.Med.Chem.,2006,49:3432.
[3] Czarna A,Popowicz G M,Pecak A,et al.High affinity interaction of the p53 peptide-analogue with human Mdm2and Mdmx[J].Cell Cycle,2009,8:1176.
[4] Pazgier M,Liu M,Zou G,et al.Structural basis for high-affinity peptide inhibition of p53interactions with MDM2and MDMX [J].Proc.Natl.Acad.Sci.USA,2009,106:4665.
[5] Phan J,Li Z,Kasprzak A,et al.Structure-based design of high affinity peptides inhibiting the interaction of p53with MDM2and MDMX[J].J.Biol.Chem.,2010,285:2174.
[6] Chen J,Liang Z,Zhang Q,et al.Insight into Interaction Mechanism of the inhibitor pDI6W with MDM2Based on Molecular Dynamics[J].J.At.
Mol.Phys.,2012,29:953(in Chinese)[陈建中,梁志强,张庆刚等.分子动力学研究抑制剂pDI6W与MDM2的相互作用机制[J].原子与分子物理学报,2012,29:953]
[7] Chen J,Zhang D,Zhang Y,et al.Computational studies of difference in binding modes of peptide and non-peptide inhibitors to MDM2/MDMX based on molecular dynamics simulations[J].Int.J.Mol.Sci.,2012,13:2176.
[8] Chen J,Zhang S,Liu X,et al.Insights into drug resistance of mutations D30N and I50V to HIV-1 protease inhibitor TMC-114:Free energy calculation and molecular dynamic simulation [J].J.Mol.Model.,2010,16:459.
[9] Wu E L,Han K L,Zhang J Z H.Selectivity of neutral/weakly basic P1group inhibitors of thrombin and trypsin by a molecular dynamics study[J].Chemistry-A European Journal,2008,14:8704.
[10] Case D A,Cheatham III T E,Darden T,et al.The Amber biomolecular simulation programs[J].J.Comput.Chem.,2005,26:1668.
[11] Case D A,Cheatham T E,Darden T,et al.The Amber biomolecular simulation programs[J].J.Comp.Chem.,2005,26:1668.
[12] Chen J,Yang M,Hu G,et al.Insights into the functional role of protonation states in the HIV-1 protease-BEA369complex:molecular dynamics sim-ulations and free energy calculations[J].J.Mol.Model.,2009,15:1245.
[13] Case D A,Darden T A,Cheatham III T E,et al.AMBER 12.San Francisco:University of California,2012.
[14] Duan Y,Wu C,Chowdhury S,et al.A pointcharge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations [J].J.Comp.Chem.,2003,24:1999.
[15] Coleman T G,Mesick H C,Darby R L.Numerical integration[J].Ann.Biomed.Eng.,1977,5:322.
[16] Cheng W,Chen J,Liang Z,et al.A computational analysis of interaction mechanisms of peptide and non-peptide inhibitors with MDMX based on molecular dynamics simulation [J].Computational and Theoretical Chemistry,2012,984:43.
[17] Cheng W Y,Liang Z Q,Zhang Q G,et al.Insight into p53-MDM2interaction based on molecular dynamics simulation and molecular mechanics[J].J.At.Mol.Phys.,2012,29:393(in Chinese)[程伟渊,梁志强,张庆刚,等.p53-MDM2 相互作用的分子力学和动力学研究[J].原子分子物理学报,2012,29:393]
[18] Chen J,Wang J,Xu B,et al.Insight into mechanism of small molecule inhibitors of the MDM2-p53 interaction:molecular dynamics simulation and free energy analysis [J].J.Mol.Graph.Model.,2011,30:46.
[19] Ding Y,Mei Y,Zhang J Z H.Quantum mechanical studies of residue-specific hydrophobic interactions in p53-MDM2 binding [J].J.Phys.Chem.B,2008,112:11396.
[20] Hu G,Wang D,Liu X,et al.A computational analysis of the binding model of MDM2 with inhibitors[J].J.Comp.Aid.Mol.Des.,2010,24:687.
[21] Lu S Y,Jiang Y J,Zou J W,et al.Molecular modeling and molecular dynamics simulation studies on pyrrolopyrimidine-based [alpha]-helix mimetic as dual inhibitors of MDM2and MDMX[J].J.Mol.Graph.Model.,2011,30:167.
