医疗器械包类产品技术审评规范探讨
2013-11-12仲志真钱虹储云高戴嗣卫汪泽朱颖峰孙旭颖
仲志真,钱虹,储云高,戴嗣卫,汪泽,朱颖峰,孙旭颖
上海市食品药品监督管理局认证审评中心,上海市,200020
医疗器械包类产品(以下简称:器械包)是指两种或两种以上器械产品(至少有一个组件应为医疗器械产品)按一定的要求组合并实现特定的医疗目的成套器械及其容器的总称。
近年来,器械包发展的速度很快,成为一种常用产品。本文主要就器械包产品的发展及现状、技术审评中发现的一些涉及产品安全有效的现象和问题以及对应的思考等几个方面进行初步地探讨。
1 器械包现状
1.1 器械包的产生原因及目前生产情况
器械包的产品形式最早是医疗机构根据某一特定的医疗目的,自行组配所需的医疗器械,由医疗机构的中心供应室进行组包、清洗、灭菌后供临床使用。随着医疗需求的不断发展,医疗机构面临的医疗压力也与日俱增,仅凭自身的供应较难满足临床的需要,逐步寻求专业机构进行定制。医疗器械生产企业看到其中很好的市场前景,进入器械包的生产、销售环节。根据医疗机构的需求,将不同的器械组合成包,或进一步灭菌后销往医疗机构。
随着医疗机构采购医疗器械越来越多采用公开招标的方式进行,出现了以招标文件需求进行组包的产品。在某种程度上刺激了医疗器械生产企业将产品打包销售的热情,加之进入该领域相对比较容易,所以有越来越多的企业加入到器械包生产企业的行列中来。这些企业中生产能力良莠不齐,对医疗器械生产质量管理和质量控制的能力差异也很大。
在我们日常的器械包的注册审评中器械包的生产方式目前主要有以下三种:
(1) 器械包内的所有组件均由申报注册的企业自身生产;
(2) 器械包内的主要组件由申报注册的企业生产,其他的组件外购;
(3) 器械包内的所有组件均由申报注册的企业外购。
第一种生产方式较少,第二种生产方式较多,也就是说申报器械包注册的企业本身就是器械包中某些组件的生产者,自身有一定的医疗器械产品生产能力。第三种生产方式,企业本身不生产器械包中的任何组件,所有的组件均向其他企业采购而得,申报器械包注册的企业仅仅是买来器械包中的组件,进行拆包装、组包,然后再将灭菌过程外包给专门的机构进行灭菌处理,自身没有任何医疗器械产品生产能力。以工作实践的经验来看,以第三种生产方式生产器械包的企业对质量管理的意识比较薄弱、对质量控制的能力一般也较弱。
1.2 器械包的种类和管理分类
随着科学技术和医疗水平的不断发展,器械包按照临床需求的变化器械包品种多、规格多。目前,常用的器械包可分为,手术器械包(例如:口腔正畸包、医用口腔包、骨科基础手术器械包、无柄髋关节安装手术器械包、普外科急救器械包等)、非手术器械包(例如:一次性使用敷料包、一次性使用气管插管包、中心静脉置管术换药包、保健盒-血压计和听诊器组合包装 等)。根据用途的不同亦可以分为护理包和工具包。护理包是指为某些特定的临床诊疗阶段提供伤口护理的器械包,比如:中心静脉置管术换药包、一次性使用敷料包等。而工具包则是为临床的特定诊疗阶段提供使用的工具,之前提及的骨科基础手术器械包、无柄髋关节安装手术器械包等应为工具包之列。根据使用的特征器械包亦可以分为重复使用和一次性使用两大类,在以上的举例中,骨科基础手术器械包、无柄髋关节安装手术器械包、普外科急救器械包、保健盒-血压计和听诊器组合包装一般是重复使用器械包;而一次性使用敷料包、一次性使用气管插管包、中心静脉置管术换药包可以为一次性使用器械包。
根据《医疗器械分类规则》(局令第15号)[1],凡器械包内含有III类医疗器械的,作为III类产品管理;只含有II类和I类医疗器械的,作为II类产品管理;只含有I类医疗器械的,作为I类产品管理。
1.3 国内器械包注册现状
依据国家局数据库统计2009年至2011年我国境内批准上市的器械包,具体情况如下:
2009年至2011年的器械包注册证数量2071个,从预期用途来看其中护理包占61.9%,工具包占36.3%,其它占1.8%。从使用特征来看,一次性使用产品占63.8%,可重复使用的产品占36.2%。按照管理类别,I类产品占51.7%、II类产品占42.6%、III类产品仅占5.7%,见图1。这三年器械包的主要生产省市情况见图2。

图1 全国器械包注册情况Fig.1 National medical devices kits registration situation
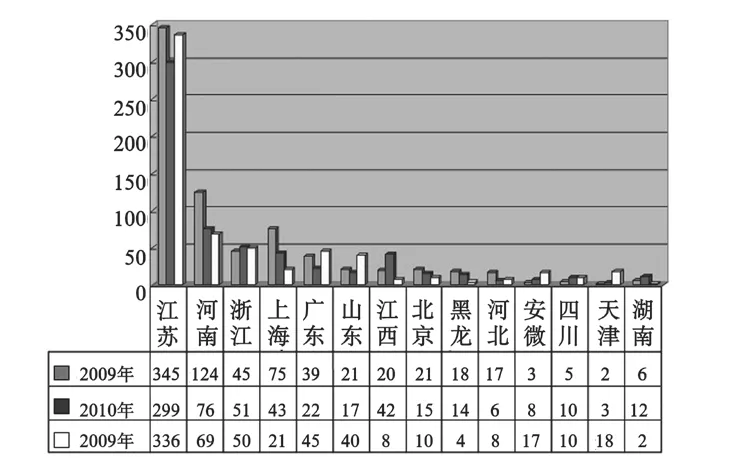
图2 主要省市器械包生产情况Fig.2 Main provinces medical devices kits production situation
由于各省市地方审批部门在对法规把握程度和对产品标准的理解方面或多或少存在着差异,加之器械包产品的覆盖的专业类别广泛,所以也存在着难于统一审评尺度的现状。
目前,上海地区有I,II类器械包生产企业约36家,I类有效注册证77张,II类有效注册证54张,III类有效注册证35张(截止到2011年12月)。近三年上海所获注册证数量见图3。
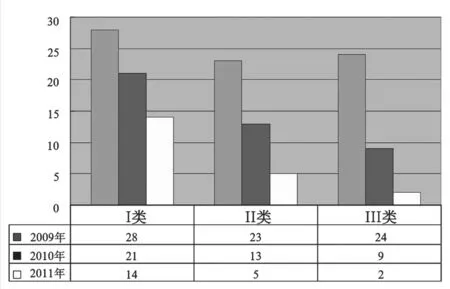
图3 上海市器械包注册情况Fig.3 Shanghai medical devices kits registration situation
从预期用途来看其中护理包占34.5%,工具包占61.8%,其它占3.7 %。从使用特征来看,一次性使用产品占38.9%,可重复使用的产品占61.1%。按照管理类别,I类产品占45.3%、II类产品占29.5%、III类产品仅占25.2%。
1.4 器械包内组件的情况
器械包中组件的情况呈现多样性,基本为以下几种:
(1) 包内组件均属医疗器械产品,均具有有效医疗器械注册证(以下简称:有证);
(2) 包内组件均属医疗器械产品,部分组件尚未经注册。
(3) 包内组件均属医疗器械产品,均未经注册;
(4) 包内组件部分属医疗器械产品,部分为非医疗器械产品,属医疗器械产品的组件均具有有效医疗器械注册证;
(5) 包内组件部分属医疗器械产品,部分为非医疗器械产品,属医疗器械产品的部分组件具有有效医疗器械注册证,部分组件尚未经注册;
(6) 包内组件部分属医疗器械产品,部分为非医疗器械产品,属医疗器械产品组件均未经注册。
2 现有法规文件和相关标准
目前,针对器械包的法律法规和相关文件不多,截止2012年年底,国家局发布了2个有关器械包产品的技术标准,YY/T0720-2009《一次性使用产包自然分娩用》[2]、YY0321.1-2009《一次性使用麻醉穿刺包》[3]。对于器械包还没有较明确的技术审评指导原则。
(1)北京市于2009年发布了《一次性使用器械包类产品技术审评规范(2009版)》[4];后又于2012年再次改版发布《一次性使用器械包(盒)产品技术审评规范(2012版)》[5]。在2012年的规范中针对导尿包、检查包、护理包、备皮包、换药包、口腔器械盒、手术敷料包等产品进行了阐述和规范。该审评规范提出对包内组件的要求如下:
①有证组件应提供医疗器械注册证;
② 未经注册的医疗器械组件应对其性能提出要求;
③其他器械应明确性能要求或提供合格证明。对含有消毒剂的产品,应有提供消毒剂卫生许可证的要求,消毒剂浓度及含量的要求,消毒效果的要求。
(2) 辽宁省在2008年5月21日对本省发布了《一次性使用无菌手术包技术指导原则》[6],该指导原则适用于以非织造布及医用脱脂纱布为主要原料加工制成的一次性使用无菌手术包(以下简称手术包),该产品于临床手术中使用。
(3) 上海市在执行医疗器械相关法规要求的基础上,对器械包的申报资料和注册技术审评提出了如下具体的要求:
①包内组件的要求
对于包内组件已取得有效医疗器械产品注册证,其主要技术要求应作主要参数写入器械包产品注册标准中。
对于包内组件包含未取得有效医疗器械产品注册证或非医疗器产品组件,需在产品标准中明确该组件的全部技术要求和相应检验方法。
② 组件的用途
有注册证产品的用途与器械包内特定用途不完全一致时,不应简单地作为有证产品在此予以认可,应由技术审评根据具体技术要求予以审查。
③有注册证组件的二次消毒或灭菌
企业首先需要确认涉及二次消毒、灭菌的组件。在组包前需要进行二次灭菌的验证。包内有注册证产品如为一次性使用的无菌产品、或有微生物控制要求的产品的,原则上不可使用半成品,应采购和使用其完整包装产品。对于部分组件拆除包装后如不改变产品使用性能的,允许拆除原包装后组包,但必须做好拆包分装记录以便溯源。
④ 组包意见及临床资料
I、II类器械包均应提交组包意见,组包意见必须包含以下内容:该器械包的用途;该器械包的组件,及各组件的特定用途;需给出“组包是否合理,临床是否适用、有效”等明确的意见。
I类器械包,所提交的“组包意见”应有2家二级甲等以上医院或者相应的专科医院的专家签字,并经其所在医院或科室盖章认可。签字专家应留有必要的联系方式以便咨询和核查。
II类器械包,所提交的“组包意见”应有2家二级甲等以上医院或者相应的专科医院的专家签字,并经医院盖章认可;若包内含有未经注册医疗器械组件的,如何提交临床资料,应执行《医疗器械注册管理办法》(局令第16号)[7]附件12中的相应要求。
3 关于注册技术审评有关问题的探讨
3.1 器械包的产品标准
在器械包的产品标准中,体现包内器械的相关信息是十分重要的。对于I、II、III类器械包来说,注册时均需要提交产品标准。在器械包产品标准中应明确列明器械包内的每一种组件的名称、采用的原材料、型号、规格、数量和医疗器械注册证的情况。
(1) 对于已单独获注册证的有证组件的状态与其注册证完整、一致的情况下,申报注册的产品标准中应明确该组件采用的是有证产品及该组件的主要技术参数、采用的原材料、具体的型号规格等信息;
(2) 对于没有取得有效医疗器械注册证的医疗器械组件,企业应在器械包的产品标准中明确该组件的结构、组成,提出该组件全部的技术要求及试验方法,若组件有国家或行业标准的,应执行相应的标准,若没有标准,则企业应科学合理的规定该组件的全部技术要求和相应的检验方法;
(3) 对于包内组件非医疗器械组件,在器械包的产品标准中明确该组件的结构、组成或应执行相关的国家(行业)标准,根据其功能性来确定相关的技术要求及试验方法。
目前在技术审评实践中发现,企业在进行器械包注册申报时,同一种用途的器械包会有多个不同的配置(不同组件的数量、同一组件不同的数量等情况)。器械包的不同的配置应在产品标准中予以明确。同时,在《医疗器械产品制造登记表》中应将所审批的器械包的每一种配置情况固定下来。以避免企业在器械包获批准上市后随意变更配置,同时监管依据也比较清晰。
3.2 器械包内有证组件的二次消毒或二次灭菌问题
器械包中有的组件在组包前已进行过一次消毒或灭菌过程,在组包后整个器械包产品送消毒或灭菌处理。对于该组件来说,其经历了两次消毒或灭菌的过程。这种二次消毒或灭菌对于该组件来说会产生怎样的影响?
对于包内有证组件为一次性使用的无菌产品、或有微生物控制要求的产品的,采购来时组件已经过消毒或灭菌过程,无论组包生产时是否进行拆除包装,企业在组包前需进行技术评估和验证以确认这种二次消毒或灭菌对于该组件使用性能、安全性的影响。在组包生产过程中,需对有证的无菌或有微生物控制的组件进行拆包装工序的,企业必须做好拆包分装记录以便溯源。
3.3 如何确认器械包内组件“有证”的问题
(1) 器械包内外购的“有证”的组件,其注册证中明确为消毒或无菌的单包装,但是器械包生产企业采购来的是非消毒或灭菌的大包装,企业进行拆包装,再组包。此种情况下的该组件是否还能够看作为有证产品,这个问题值得我们思考。
在上述情况下,经过拆大包装或中包装的组件仅仅是半成品而已,与有证产品比较,缺少产品包装过程和一个灭菌或消毒的过程,该组件不能被简单的视作为有证组件。器械包生产企业应对该半成品组件的生物负载进行测量,并将之列入评估消毒或灭菌时需要考虑的参数;同时对于增加的生物负载应考虑该半成品组件在消毒或灭菌后可能增加的热原等不良因素。
(2) 器械包中“有证”的组件,其在器械包中起到的作用与该组件的注册证中所批示的临床预期用途不一致时,该组件是否还能够被视作为“有证”组件?
当器械包中“有证”的组件,在器械包中起到的临床用途与该组件的注册证中所批示的临床预期用途不一致时,企业不应将该组件作为“有证”产品组件。技术审评时需要按照其实际的临床用途对其进行技术评价,并应在器械包标准中规定该组件与其临床用途相适应的全部技术指标和相应的试验方法。
3.4 器械包证后包内组件的替换
当器械包在获得注册批准后,若企业对器械包中某些医疗器械组件的采购供方发生变化企业是否需要向医疗器械主管部门提交备案说明?是否会涉及《医疗器械注册管理办法》(局令第16号)中有关重新注册条款(第三十四条)的规定?
以上的这种情况在实际中屡见不鲜,但是不宜简单的认为仅是器械包组件的供方发生改变。这需要企业进行技术评估替代组件在原材料、产品特征(结构及组成等)、主要技术参数、临床用途等方面是否能够达到原先组件的水平。若器械包中的起主要临床作用的有证组件发生替代的,则需要企业对其安全性指标和主要的有效性指标进行验证,确认后方可进行替换。
3.5 器械包中直接接触患者非医疗器械组件的审评要点
对于器械包内直接接触患者的非医疗器械组件,如滑石粉、口杯等,它们的安全性如何考虑?
上述组件,因直接接触患者,企业需要对该组件的毒性重点评价。企业应在产品标准中明确该组件所采用的原材料、物理和化学特征,并应根据这些组件与人体接触的方式、接触的部位、接触的时间,对该组件的生物相容性作出全面的评价。
3.6 器械包中含消毒剂组件的审评要点
在器械包中含消毒剂的组件不在少数,例如:碘伏棉球、酒精棉棒、酒精棉球等。对于消毒剂我国有明确的规定,消毒剂应取得《国产消毒剂和消毒器械卫生许可批件》。在此类器械包的注册申报资料中,器械包生产企业必须提交该消毒剂的相关资质证明。技术审评时应了解器械包中消毒剂是否取得这样的许可。同时应注意消毒剂的用途、使用的范围、方法和注意事项等内容应与《国产消毒剂和消毒器械卫生许可批件》批准的内容一致进行审核。
3.7 具有医疗器械注册证的器械包中的某一组件是否可以作为“独立有证”的“产品”
当一个器械包在获得医疗器械注册批准后,其中的某一组件是否能够作为“有证”的组件被包入其他器械包中?
一个器械包获得医疗器械注册证时,原先包内的非有证组件的用途、使用方法等等,是依据该组件在整个器械包中的使用过程中的作用进行技术审查的,该组件在另一种组包形式下,临床用途和使用方法等可能与原有证的器械包不完全相同,因此不能因为原整个器械包已经注册而认为某一组件就是“有证”。当该组件作为其他器械包的组件时,应该仍然被视为无证的医疗器械组件进行技术评价。
3.8 器械包的有效期
器械包中的组件可能会有自己的有效期,比如:消毒剂等,对于如何确定器械包的有效期显然是个比较棘手的问题。目前企业没有过多的考虑此类问题,器械包生产企业需要对整个器械包有效期进行验证;具体到每种组件都需要考虑有效期的验证,最后以组件最短有效期的作为器械包的有效期。
3.9 无任何包内组件生产能力的企业从事“器械包”组包生产
在现实情况中有些器械包生产企业用于组包的组件全部外购,申报器械包注册的企业仅对买来的组件进行拆包装,组包,或再将灭菌过程外包给专门的机构进行灭菌处理。此种“器械包生产企业”的生产方式能否能理解为“生产”?
这些企业的生产方式与其说是“生产”,还不如说其更接近于“经营”行为。企业自身对产品的理解、对产品的质量管理的意识和质量控制的能力均很薄弱,企业对生产过程中对可能产生的风险认识也不足。这给器械包产品本身会带来很多的不确定性。但目前在法规层面对这类生产企业没有很清晰的要求,有待今后法律法规的完善。
3.10 器械包类产品的临床资料问题
根据《医疗器械注册管理办法》(局令第16号)的要求,I类器械包不需要提交临床资料。II类器械包需要提交临床资料。但是法规中找不到对于器械包这一特殊产品的具体要求。
器械包内的组件在绝大部分情况下均是常见的器械,器械包本身的临床用途是这些组件协同配合的结果,所以器械包本身的组包是否合理是临床考虑的关键。因此,无论是I类器械包还是II类器械包都应在注册申请时提交由医疗机构出具的组包意见。若器械包内的II类医疗器械组件为“无证”产品,且不在国家食品药品监督管理局出台的临床豁免目录中,则需要企业提供该组件的临床资料或整个器械包的临床资料。
4 结语
在医疗器械的注册技术审评中,器械包产品与单一的医疗器械在审评的原则和思路上并没有很大的区别,但是器械包产品在具体的技术审评尺度把握上容易出现分歧。笔者在实际的工作中遇到了一些问题希望能够从现有的法规层面上得到解决之道,但是结果差强人意。所以这篇文章是基于将我们在实际工作中的困惑和思考呈现出来,如何在安全、有效的两大原则基础上使得器械包的注册技术审评更加完善,使得器械包在上市后更加规范、有序。由于作者的能力有限,作此文章完全希望能引起从事相关工作的同行们的注意,以此抛砖引玉,使器械包注册技术审评工作得以更好的开展。
[1]国家药品监督管理局.医疗器械分类规则(局令第15号)[S].2000.
[2]国家食品药品监督管理局.YY/T 0720-2009 一次性使用产包 自然分娩用[S].
[3] 国家食品药品监督管理局.YY 0321.1-2009 一次性使用麻醉穿刺包[S].
[4] 北京市药品监督管理局.一次性使用器械包类产品技术审评规范 2009版[S].
[5] 北京市药品监督管理局.一次性使用器械包(盒)产品技术审评规范(2012版)[S].
[6] 辽宁省食品药品监督管理局技术审评中心.一次性使用无菌手术包技术指导原则[S].2008.
[7] 国家食品药品监督管理局.医疗器械注册管理办法(局令第16号)[S].2004.
