匍枝根霉TP-02 内切葡聚糖酶基因eg2 的克隆表达及功能分析*
2013-10-30汤斌张莹莹杨亚平
汤斌,张莹莹,杨亚平
(安徽工程大学 微生物发酵安徽省工程技术研究中心,安徽 芜湖,241000)
木质纤维素在地球上的分布十分广泛且蕴藏量丰富,是最廉价的可再生性生物质能源[1]。纤维素酶为多组分酶系,主要包括内切葡聚糖酶(endoglucanases,EG)、外 切 葡 聚 糖 酶(cellobiohydralases,CBH)和β-葡萄糖苷酶(β-glucosidases,BG),三者通过协同作用,分解纤维素获得各种寡糖以及纤维二糖,最终水解生成葡萄糖等单糖,继而可运用于工业生产制取酒精、医药用品、食品以及其它化工原料等,具有良好的应用前景[2-3]。其中,内切葡聚糖酶主要对纤维素内部的非结晶区起作用,通过随机切割斩断β-1,4-糖苷键,释放出长度不等的短链低聚糖,从而在纤维素的降解过程中起着至关重要的作用[4-5]。目前,如何提高内切葡聚糖酶活力及产量已成为了研究的一大热点。国内外对内切葡聚糖酶的研究主要集中于基因克隆表达和产酶条件优化,现已从多种真菌如里氏木霉、斜卧青霉、米根霉等克隆得到内切葡聚糖酶基因,并成功构建基因工程菌[6-8]。而对于内切葡聚糖酶结构与功能的研究,尤其是其催化过程中关键基团的报道还较少。
纤维素酶分子通常由纤维素结合域(CBD)、催化结构域(CD)及长短各异的连接片段(linker)组成[9-10]。CBD 由各种纤维素结合模块(CBM)组成,其功能为吸附于纤维素表面并将CD 呈递到底物之上,以疏解结晶纤维素的结构[11];CD 呈现出对底物的特异性,可以独立发挥催化作用[12]。本文从匍枝根霉TP-02 的cDNA 文库中克隆得到eg2 基因,并对其基因产物EGⅡ的CBM 及CD 的结构功能进行分析,在此基础上确定参与酶解过程的重要残基,并对其进行突变分析,以期通过基因修饰的手段提高内切葡聚糖酶的催化活力。
1 材料与方法
1.1 菌株、质粒和引物
匍枝根霉TP-02 菌株由本实验室从黄山生态林的腐木中分离并保存;大肠杆菌(Escherichia coli)BL21 菌株和pET28a 质粒均由本实验室保存。引物均由上海生工合成。
1.2 试剂及培养基
Trizol 试剂、mRNA 抽提试剂盒、PCR 产物纯化试剂盒、质粒抽提试剂盒均购自上海生工,cDNA 反转录试剂盒、T4 DNA 连接酶及各种限制性内切酶均购于TaKaRa 公司(大连)。
诱导培养基:CMC-Na 0.4%,KH2PO40.2%,(NH4)2SO40.14%,尿素、MgSO4和CaCl2各0.03%,微量元素(FeSO45 mg/L,MnSO41.5 mg/L,ZnSO41.4 mg/L,CoCl22.0 mg/L,蛋白胨0.75 mg/L,酵母膏0.25 mg/L)。筛选培养基(CMC-Na):CMC-Na 1.5%,(NH4)2SO40.3%,KH2PO40.1%,MgSO40.05%,琼脂2%。含刚果红筛选培养基:配制10 mg/mL 刚果红溶液,灭菌后按1 mL/200 mL 的比例加到CMC-Na 筛选培养基中,混匀后倒平板。
PDA 培养基及LB 培养基配方参见分子克隆指南。
1.3 PCR 扩增eg2 基因
将匍枝根霉TP-02 扩大培养后转入诱导培养基中,诱导72 h 后离心收集菌体,进行冷冻干燥。取100 mg 菌丝体在预冷的研钵中快速研磨,利用Trizol试剂提取总RNA,并根据试剂盒法纯化mRNA,以其为模板利用cDNA 试剂盒反转录合成cDNA 第一链。设计引物扩增目的基因,其上下游引物分别为F1 和R1(下划线所示为酶切位点):
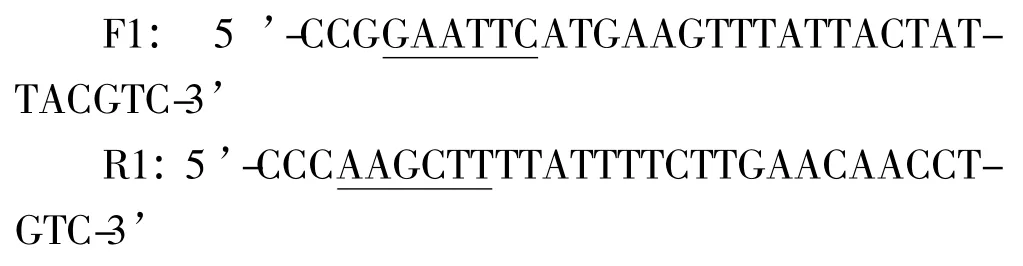
反应条件为:95℃预变性5 min;94℃变性30 s,56℃退火30 s,72℃延伸75 s,共30 个循环;72℃终延伸10 min。
1.4 重组质粒的构建及阳性克隆筛选
将纯化的PCR 产物和pET28a 质粒分别经EcoRⅠ和Hind Ⅲ双酶切后连接转化,构建表达质粒pET28a-eg2。将重组质粒导入BL21 中,加入IPTG 至终浓度为1 mmol/L 涂布于含Kan+的LB 平板上,将形成的单菌落依次点种于含刚果红的CMC-Na 筛选培养基上,并加入IPTG,37℃培养至长出单菌落,挑取透明圈较大的单菌落保种,用于酶切验证、测序及发酵性能的研究。
1.5 重组菌株的诱导表达
挑取阳性克隆单菌落,接种于LB 液体培养基中,37℃,200 r/min 振荡培养至OD600达0.8 左右,加入100 mmol/L 的IPTG 使其终浓度为1 mmol/L,进行诱导表达。每3 h 取样,分别取1 mL 菌液,5 000 r/min 离心10 min,去上清液收集菌体,用0.05 mol/L柠檬酸缓冲液(pH 5.0)重悬细胞,-80℃反复冻融进行破胞,5 000 r/min 离心30 min,取上清液测酶活,并进行SDS 聚丙烯酰胺凝胶电泳(5%浓缩胶,12%分离胶)。
1.6 酶活测定
采用DNS 法测定内切葡聚糖酶的活力[13]。酶活定义:在酶的催化下,每分钟水解CMC-Na 生成1 μmol 葡萄糖所需的酶量为1 个酶活力国际单位IU。
1.7 eg2 基因编码蛋白质的生物信息学分析
利用PROSITE 数 据 库(http://prosite. expasy.org/)预测EGII 的主要功能位点,并借助CDD 数据库(http://www. ncbi. nlm. nih. gov/Structure/cdd/wrpsb.cgi)对EGII 氨基酸序列的保守结构域进行分析。利用ClustalX1.8 软件对EGII 各保守结构域及其相应的同源序列进行多序列比对,并用BioEdit 软件进行分析。利用Phyre2 在线分析引擎(http://www.sbg.bio.ic.ac.uk/ ~phyre/index.cgi)完成EGII三级结构的同源建模,并用Discovery Studio 2.5 软件进行分析。
1.8 eg2 的基因修饰
根据eg2 生物信息学分析结果,确定突变位点并设计突变组。以pET28a-eg2 为模板,利用重叠PCR完成eg2 基因的修饰。各突变组如表1 所示;各组引物见表2,下划线为突变位点,其中A1,B1 参与各突变组的PCR 过程。PCR 条件如下:95℃预变性5 min;95℃变性30 s,54℃/55℃退火45 s,72℃延伸50 s,共22 ~25 个循环;72℃终延伸10 min。将突变体酶切后与pET28a 连接,测序并转染大肠杆菌BL21,测定各突变株的酶活。
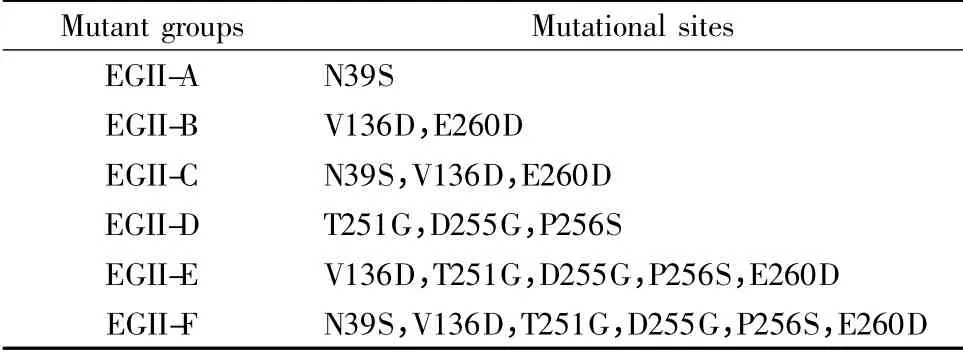
表1 突变组设计Tabel 1 Design of mutant groups
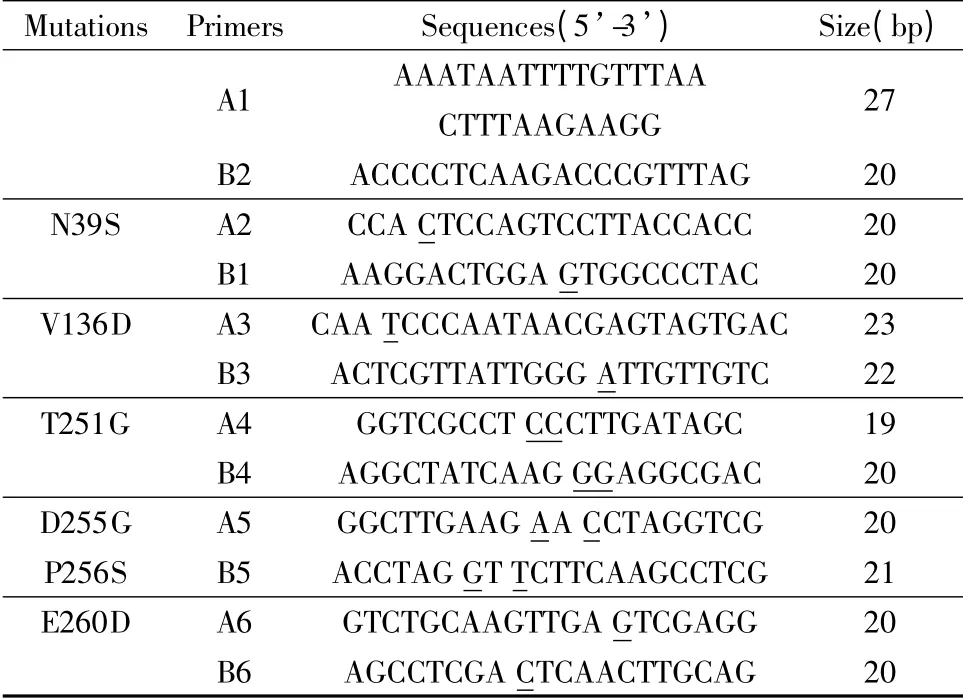
表2 重叠PCR 的引物序列Tabel 2 The sequences of primers for overlapping PCR
2 结果
2.1 eg2 基因的克隆
提取得到高质量的总RNA,并从中纯化获得mRNA,经逆转录酶作用,成功合成匍枝根霉TP-02的cDNA 单链,并从中克隆得到一段954 bp 的新型内切聚糖酶基因,称为eg2 基因,将其上传至Gen-Bank,获取登录号为JX315341。双酶切验证结果见图1。
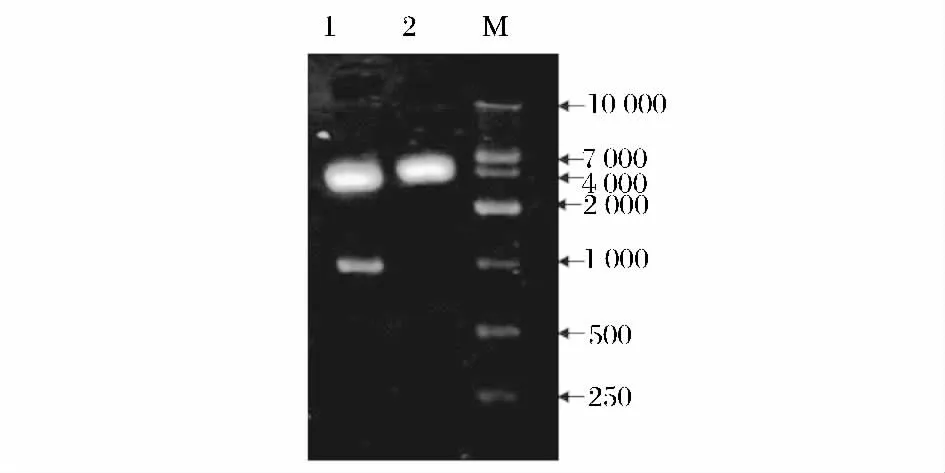
图1 重组质粒双酶切验证Fig.1 Identification of recombinant plasmid
2.2 阳性克隆筛选
刚果红与大分子多糖可牢固结合,随着多糖分子量的变化,刚果红结合程度就会发生变化,导致颜色从深红变为淡黄。随着菌落的长大,CMC 被不断降解为寡糖,在菌落周围就会形成透明圈,其大小由CMC 酶的降解能力所决定。透明圈越大,酶活越高。故而能够快速地分辨出阳性克隆。图2 所示为阳性克隆所形成的透明圈,空白对照A 在筛选平板上无法正常生长,未形成透明圈。

图2 刚果红染色结果Fig.2 The result of Congo red stain
2.3 EGII 功能位点及保守结构域预测
经PROSITE 分析,EGII 序列中含有1 个碳水化合物结合区(CBM1),该功能区的位置为氨基序列的25 ~61 位。CDD 数据库的分析结果显示,该蛋白质归属于糖基水解酶第45 家族(Glycoside Hydrolase Family 45),包含2 个GH45 家族的特征序列,并含有1 个典型的真菌纤维素结合区域即CBM1,该纤维素结合区域由4 个半胱氨酸构成,与PROSITE 预测结果一致。
2.4 EGII 氨基酸序列分析结果
2.4.1 CBM 模块的比对分析
利用ClustalX 对EGII 的CBM1 序列进行同源比对,发现其在第15 位(即全序列的39 位)上的丝氨酸保守残基被天冬酰胺所取代,该残基参与CBM 亲水性平面的形成[14],故该位点的突变可能会导致纤维素酶结合区域的结构改变,而对其酶活产生影响。以此确定CBM 的突变位点为N39S。
2.4.2 催化结构域GH45 特征序列分析结果
内切葡聚糖酶的催化区域通常包含两个天冬氨酸催化残基[1,15],通过序列比对发现EGII 中相对应的位置为136 位和260 位,均被其他残基所取代,故确定其突变位点为V136D 和E260D。此外,文献中报道在催化中心的一侧存在一段loop 环,该环能增强催化基团周围环境的疏水性,阻止除水分子外的各种亲核试剂进入活性部位[15]。经序列比对后,确定该loop 环的突变位点为T251G、D255G 和P256S。利用Phyre2 完成EGII 的催化区GH45 及其突变体的三维建模,结果如图3 所示。催化残基V136D 及E260D 的突变形成拉近了两者的距离,以其为中心的活性区域发生改变。此外,对位于V136 和E260 中间的一段loop 环进行改造,T251G、D255G 及P256S的突变形成使得该loop 环移至活性中心一侧,同时改变催化残基D260 的空间位置。
2.5 发酵性能实验
将含eg2 基因的大肠杆菌重组菌及各突变株接到LB 液体培养基中进行发酵培养,每隔3 h 取样测CMC 酶活,结果如图4 所示。EGⅡ-A 至EGⅡ-F 的最高酶活分别为0.897 IU/mL,1.114 IU/mL,1.286 IU/mL,0.868 IU/mL,1.215 IU/mL 和1.321 IU/mL。各突变株的酶活均高于原始菌株,其中EGⅡ-F 的酶活比EGⅡ(0.723 IU/mL)提高了82.7%。含CBM1突变位点N39S 的突变株EGⅡ-A、EGⅡ-B 和EGⅡ-F比其他重组菌更早达到峰值,而所有含V136D 及E260D 的突变株均具有较高的酶活。单独对loop 环进行突变的重组菌EGⅡ-C 酶活较低。
2.6 SDS-PAGE
取以上酶活达到峰值时的发酵液进行SDS 聚丙烯酰胺凝胶电泳,以未诱导的EGⅡ为空白对照,结果如图5 所示。表达条带的分子量大小约为45 kDa,与理论值相符。其中,EGⅡ-E 和EGⅡ-F 的表达条带较粗,表达量相对较高。
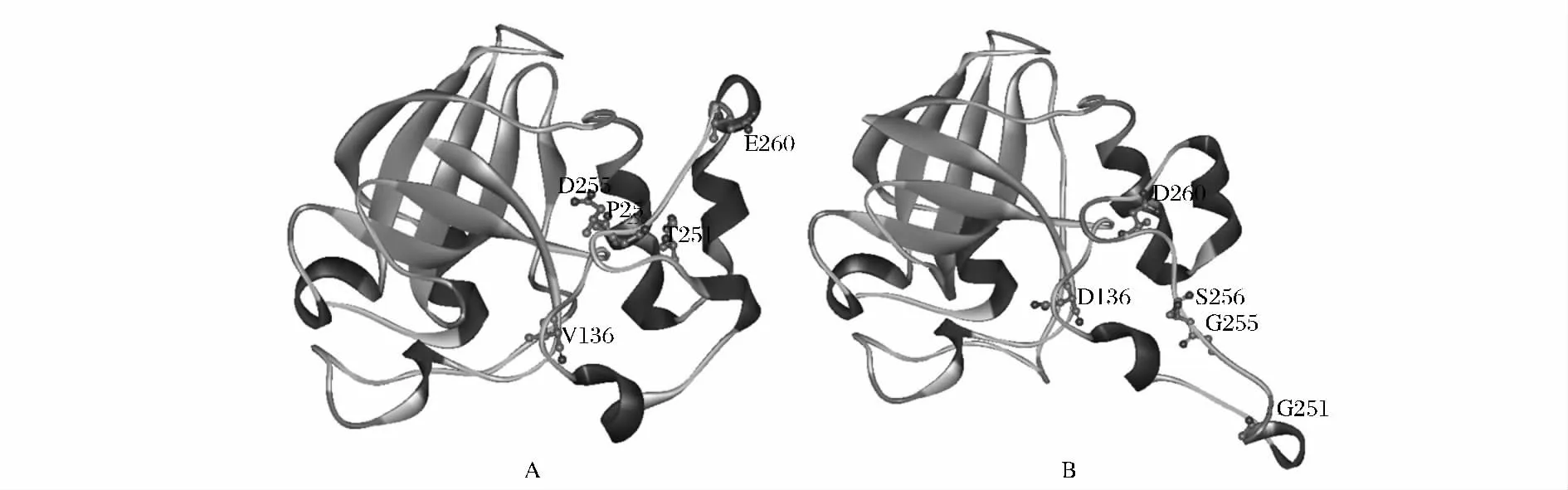
图3 GH45 三级结构模型Fig.3 Tertiary structure of GH45 domain
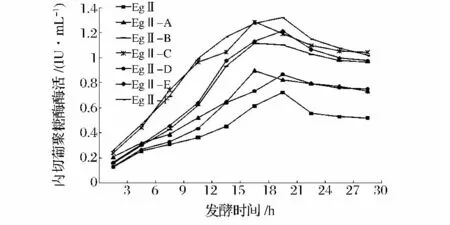
图4 重组菌的发酵特性Fig.4 Fermentation characteristics of different recombinant E. coli BL21
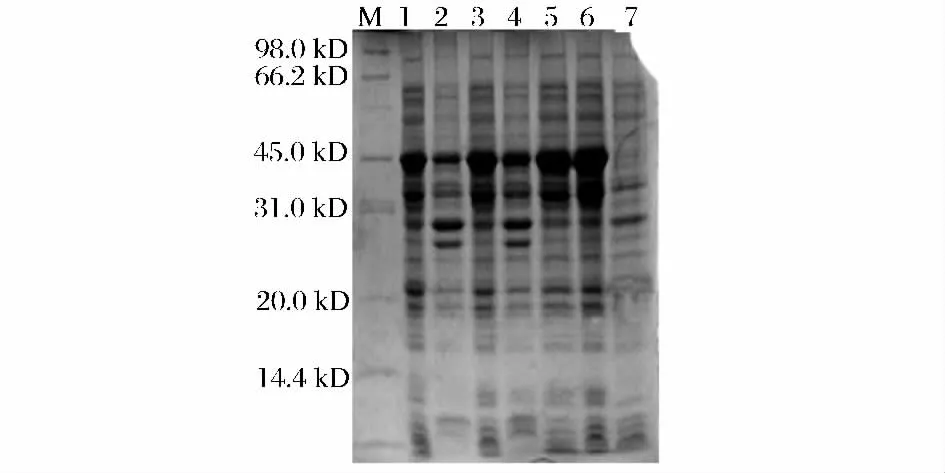
图5 SDS-PAGE 结果Fig.5 Results of SDS-PAGE.
3 讨论
本文成功克隆了匍枝根霉的内切葡聚糖酶基因,并实现了其在大肠杆菌中的表达,对其结构功能进行了初步探讨,发现纤维素结合模块CBM1 和催化结构域GH45 区在纤维素降解过程中起着关键作用。突变分析结果显示,N39S 突变的形成可能会对CBM1与纤维素结合能力产生影响,从而改变内切葡聚糖酶的水解效率。V136D 及E260D 两个催化位点的突变形成对酶活有明显的提高作用,而单独对loop 环进行突变只能对酶活产生微弱的影响,即在酶解过程中该loop 环只起辅助作用。然而,对于CBM1 和GH45结构域的具体作用机理还不甚明了,后续研究将着眼于纤维素酶的水解机制,及各保守残基在催化过程中所扮演的角色。
[1] Gowen CM,Fong SS. Exploring biodiversity for cellulosic biofuel production[J]. Chemistry & Biodiversity,2010,7(5):1 086 -1 097.
[2] Takashima S,Ohno M,Hidaka M,et al. Correlation between cellulose binding and activity of cellulose-binding domain mutants ofHumicola griseacellobiohydrolase Ⅰ[J].FEBS Letters,2007,581(30):5 891 -5 896.
[3] 韦小敏. 斜卧青霉胞外蛋白质组学分析与纤维素酶合成调控机制研究[D]. 山东大学,2011:1 -2.
[4] Nakazawa H,Okada K,Onodera T,et al. Directed evolution of endoglucanaseⅢ(Cel12A)fromTrichoderma reesei[J]. Applied Microbiology Biotechnology,2009,83(4):649 -657.
[5] 郑海英,黄平,蔡少丽,等. 特异腐质霉内切葡聚糖酶Ⅱ基因在毕赤酵母中的表达及酶学性质[J]. 微生物学通报,2012,39(2):145 -153.
[6] Samanta S,Basu A,Halder UC,et al. Characterization ofTrichoderma reeseiendoglucanase Ⅱ expressed heterologously in Pichia pastoris for better bionfinishing and biostoning[J]. J Microbiol,2012,50(3):518 -525.
[7] 刘韫滔,韩学凤,罗泽宇,等. 斜卧青霉L-06 内切葡聚糖酶Ⅰ基因的克隆与表达[J]. 微生物学通报,2012,39(5):696 -701.
[8] Moriya T,Murashima K,Nakane A,et al. Molecular cloning of endo-beta-D-1,4-glucanase genes,rce1,rce2,and rce3,fromRhizopus oryzae[J]. J Bacteriol,2003,185(5):1 749 -1 756.
[9] Hirvonen M,Papageorgiou AC. Crystal structure of a family 45 endoglucanase fromMelanocarpus albomyces:mechanistic implications based on the free and cellobiose-bound forms[J]. Journal of Molecular Biology,2003,29(3):403 -410.
[10] 方诩,秦玉琪,李雪芝,等. 纤维素酶与木质纤维素生物降解转化的研究进展[J]. 生物工程学报,2010,26(7):864 -869.
[11] Carrard G,Koivula A,Soderlund H,et al. Cellulosebinding domains promote hydrolysis of different sites on crystalline cellulose[J]. Proc Natl Acad Sci USA,2000,97(19):10 342 -10 347.
[12] Warner CD,Camci-Unal G,Pohl NLB,et al. Substrate binding by the catalytic domain and carbohydrate binding module ofRuminococcus flavefaciensFD-1 xyloglucanase/endoglucanase[J]. Industrial & Engineering Chemistry Research,2013(2):30 -36.
[13] 陈红漫,张欣,李艳秋,等. 绿色木霉葡聚糖内切酶(EGⅠ)基因克隆及在酿酒酵母中的表达[J]. 食品与发酵工业,2012,38(6):48 -52.
[14] Kraulis PJ,Clore GM,Nilges M,et al. Determination of the three-dimensional solution structure of the C-terminal domain of cellobiohydrolase I fromTrichoderma reesi. A study using nuclear magnetic resonance and hybrid distance geometry-dynamical simulated annealing[J]. Biochemistry,1989,28(18):7 241 -7 257.
[15] Valjakka J,Rouvinen J. Structure of 20K endoglucanase fromMelanocarpus albomycesat 1.8 resolution[J]. Biological Crystallography,2003,59:765 -768.
