植物转录因子最新研究方法
2013-10-27王传琦孔稳稳李晶
王传琦,孔稳稳,李晶
东北农业大学 生命科学学院,黑龙江 哈尔滨 150030
转录因子(transcription factor,TF)又称反式作用因子,是直接或间接与基因启动子区域中顺式作用元件发生特异性相互作用,并对基因转录的起始进行调控的一类蛋白质。转录因子一般具有4个功能结构域,即DNA结合区(DNA-binding domain,DBD)、寡聚化位点区(oligomerization site,OS)、核定位信号区(nuclear location signal,NLS)和转录调控区(transcription regulation domain,TRD)。DNA 结合区是转录因子识别并结合于各种顺式因子的一段氨基酸序列,可与靶位点专一性结合;寡聚化位点区是不同转录因子间发生相互作用的功能域;核定位信号可将转录因子定位于细胞核内的某些区域;转录调控区决定着转录因子对靶基因的调控特征,可分为转录激活域(activation domain,AD)和转录抑制域(repression domain,RD),对靶基因的转录起激活或抑制作用。
自植物中最早的玉米转录因子被报道以来[1],已发现植物中存在大量转录因子。根据模式植物拟南芥信息资源库TAIR提供的数据分析,拟南芥基因组中包含27 235个编码蛋白的基因,其中2000个以上编码转录因子,约占基因总数的5%~10%,明显高于果蝇(4.7%)和线虫(3.6%)[2]。转录因子的数量和种类繁多,表明了植物转录调控的特殊性和复杂性。目前已从高等植物中分离鉴定出数百种转录因子,有些转录因子参与调控植物细胞和组织的形成,是植物形态建成的关键调节因子,如bHLH转录因子家族[3-4]。大量转录因子与植物抗逆性密切相关,可调控植物体感受干旱、高盐、低温和病原等信号相关基因的表达,在植物抗逆反应中发挥重要作用,如bZIP类转录因子[5-6]。转录因子还可对植物次生代谢产物的合成和分解进行调节,影响次生代谢产物的合成、分解及时空分布,如MYB转录因子家族[7-8]。
由于转录因子在植物生长发育、形态建成及对外界环境变化的反应中起重要的作用,在植物基因表达调控研究中转录因子一直备受关注。我们结合近年来植物转录因子的研究进展,归纳分析了高等植物转录因子研究的主要策略和最新的技术方法,可为植物转录因子的鉴定、功能分析及靶基因研究提供理论和技术上的参考。
1 生物信息学分析
随着生物信息学的迅速发展,各种分子生物学数据库提供了大量核苷酸和氨基酸序列,对这些序列进行分析和计算,已成为发现新基因和预测蛋白质结构最快捷有效的方法。常用的大型生物学数据库NCBI、EBI和DDBJ等都能提供植物转录因子的相关数据。另外还有许多转录因子的专用数据库,如 TRANSFAC(http://www.gene-regulation.com/pub/databases.html#transfac)和 PlnTFDB(http://plntfdb.bio.uni-potsdam.de/v3.0/)等。TRANSFAC是关于转录因子在基因启动子上结合位点的数据库[9],而PlnTFDB是系统收录植物转录因子的数据库[10-11]。模式植物通常有自己专属的转录因子数据库,如拟南芥转录因子数据库RARTF(http://rarge.gsc.riken.jp/rartf/)、AGRIS(http://arabidopsis.med.ohio-state.edu/AtTFDB/)和 DATF(http://datf.cbi.pku.edu.cn/),每个数据库都有自己的分类方式,并对这些转录因子的功能结构和作用位点都进行了详细的描述[12]。水稻也有自己专门的转录因子数据库DRTF(http://drtf.cbi.pku.edu.cn/)[13]。这些数据库对植物转录因子的研究有重要的指导作用。
1.1 转录因子保守结构域的分析
大多数转录因子的序列具有保守性,这些保守结构域(conserved domains,CD)使得用生物信息学方法进行转录因子的预测和功能鉴定成为可能。早期研究中常利用已知转录因子的核酸序列作为探针,与cDNA文库进行杂交来筛选转录因子,这种方法耗时、费力。现在可以利用生物信息学来研究一个未知的序列是否属于转录因子。DBD是转录因子最主要的保守区域,其次还有AD或RD。在漫长的进化过程中,这些区域的氨基酸序列变化都很小,确保了转录因子功能的准确行使。可见保守区是确定一种蛋白质是否是转录因子并鉴定其功能和类型的关键。通常一个转录因子含有1个DBD和1个AD或RD。只含有1个DBD的转录因子一般具有转录抑制活性,如CPC和TRY[14]。也有些转录因子含有2个DBD,如RAV1家族成员,同时含有AP2-ERF和B3等2个DBD[15]。目前已有很多转录因子保守结构域数据库及分析软件,比较常用的如欧洲生物信息研究院(EBI)提供的 InterProScan(http://www.ebi.ac.uk/Tools/interProScan/)[16]。如果是在一系列蛋白质中寻找已知或未知的转录因子CD,则可利用MEME数 据 库 (http://meme.sdsc.edu/meme/intro.html)[17]。SALAD(http://salad.dna.affrc.go.jp/salad/en/)是 专 门针对植物蛋白质建立的数据库,包含了MEME中的CD数据,同时提供了各种分析软件和工具。除了CD以外,在其他区域具有较高同源性的转录因子功能上冗余的可能性也相对较大,因此可利用NCBI数据库的BLAST对多个转录因子的CD及其他序列进行同源性分析,以便更准确地预测这些转录因子的功能。
1.2 转录因子亚细胞定位的分析
转录因子只有在细胞核中才能发挥其功能,所以对候选转录因子进行生物信息学的亚细胞定位分析十分重要。某些转录因子的核定位信号肽可使其定位于细胞核内。根据氨基酸序列可以分析和预测蛋白质的亚细胞定位。SUBALL数据库(http://www.plantenergy.uwa.edu.au/suba2/)[18]可 提 供 大 量 蛋 白 质定位的实验数据,并通过10种不同的计算程序来计算和预测蛋白质的亚细胞定位。还有一些其他的计算软件如 TargetP(http://www.cbs.dtu.dk/services/Tar⁃getP/)[19]、SubLoc(http://bioinfo.tsinghua.edu.cn/Sub⁃Loc/)[20]和 WoLF PSORT(http://wolfpsort.org/)[21]等 。这些数据库和软件给植物转录因子亚细胞定位研究提供了极大的便利,但有时不同计算方法可能会得出不同的预测结果,最终的结论还必须通过实验来验证。
1.3 转录因子的表达及调控
转录因子对下游靶基因的表达起调控作用,而转录因子基因自身的表达也可能受到其他调节因子的调控。和其他基因一样,转录因子基因的编码区上游含有各种顺式调控元件来接受调控因子的作用。这些顺式作用元件的相关数据可通过AGRIS ATCISDB(http://arabidopsis.med.ohio-state.edu/Atcis⁃DB)和 PLACE(http://www.dna.affrc.go.jp/PLACE/)获得。
microRNA(miRNA)是影响转录因子基因表达的重要因素,它是一种长18~24 nt的非编码小RNA,可以在转录及转录后水平对基因的表达进行调节。研究表明,拟南芥已知miRNA的候选靶基因中35%编码转录因子,这些miRNA在拟南芥生长发育和形态建成中起至关重要的作用[22]。除了miRNA外,其他形式的小RNA也参与转录因子的调控。拟南芥的ASRP数据库(http://asrp.cgrb.oregonstate.edu/)提供包括siRNA、ta-siRNA及miRNA在内的所有小RNA信息,是研究转录因子自身表达调控的有力工具。
许多转录因子并不能独自行使功能,它们需要与其他蛋白形成复合体或被激酶磷酸化激活,如MADS家族[18]和MYB家族[23]通常会形成蛋白质复合体。EBI(http://www.ebi.ac.uk/)提供了大量经过实验验证或计算机预测的蛋白质-蛋白质相互作用的数据。模式植物拟南芥蛋白质间相互作用的信息还可从 TAIR(http://www.arabidopsis.org/)和 AtPID(http://www.megabionet.org/atpid/webfile/)等数据库中获得。
2 瞬间转化分析
转录因子可通过激活或抑制实现对下游靶基因转录的调控,利用瞬间转化技术可对转录因子的调控特性进行分析[24]。具体方法是构建2种植物表达载体(图1),一种是效应载体,可高水平表达待研究转录因子和一个外源DBD的融合基因,外源DBD可以采用来源于酵母的GAL4DB。另一种是报告载体,有2种不同情况:一种是用于检测转录因子是否具有激活活性的报告载体A(active),载体包含一个特异性启动子和一个报告基因,该启动子须含有可以与效应载体中的DBD特异性结合的顺式作用元件。当效应载体和报告载体同时转入植物细胞后,效应载体中的DBD与报告载体中启动子特异性结合,如果报告基因表达量增加,则说明该转录因子具有激活活性。另一种是用于检测转录因子是否具有抑制活性的报告载体R(repress),载体须含有一个组成型强启动子如花椰菜花叶病毒(CaMV)35S启动子和一段与效应载体中的DBD特异性结合的顺式作用元件及报告基因。将其与效应载体同时转入植物细胞后,如果报告基因表达量降低,则说明该转录因子具有抑制活性。报告基因可以采用绿色荧光蛋白(GFP)、β-葡萄糖苷酸酶(GUS)或萤光素酶(LUC)等基因。瞬间转化可采用基因枪法、电转化法、PGE处理法及农杆菌介导法,在几小时或几天后可观察到报告基因的表达情况[25-26]。其中,农杆菌介导的烟草叶片瞬间转化经济、便捷、高效。
另外,瞬间转化分析也可用于验证转录因子与靶基因启动子之间的识别特性。在待测靶基因的启动子序列下游加上报告基因,和转录因子一起转入植物组织,观察报告基因的表达,如果报告基因的表达发生预期改变,则可确定两者之间存在特异性的识别。
3 突变体表型分析
鉴定转录因子生理功能最直接的方法是观察转录因子基因表达发生改变时植物的表型。通常利用基因过量表达和基因功能缺失突变体,观察植物外部形态、生理代谢和对某些特殊外界环境的反应是否发生改变,分析该转录因子的功能。
3.1 基因功能增加表型分析
基因功能增加可采用超表达法,即在目的转录因子基因前加入一个组成型强启动子如CaMV 35S启动子,使转录因子大量表达;如果待研究转录因子具有转录激活活性,则还可将其与一个外源的转录激活域相融合,如来自疱疹病毒的VP16-AD,使其激活活性增强,从而促进下游基因的大量表达,相当于将转录因子基因过量表达。2006年,Ichikawa等在模式植物拟南芥中建立了基因过量表达的高通量分析系统FOX-hunting system(Full-length cDNA Over-eXpressing gene hunting system)[27]。 FOX-hunting system首先创建待研究植物全长cDNA文库,将均一化的全长cDNA在拟南芥中过量表达,筛选具有显著表型变化的转基因植株,再通过基因组PCR分离鉴定引起表型变化的基因,进一步确定其功能[27-29]。拟南芥的FOX转基因株系目前可通过RIKEN Plant Science Center获得(http://pfgweb.psc.riken.jp/index.html)。Fujita等在研究大量转录因子时采用小规模的FOX-hunting system方法,将43种逆境诱导的转录因子全长cDNA混合转入拟南芥,在筛选到的耐盐株系中鉴定出转录因子AtbZIP60的逆境信号转导功能[30]。
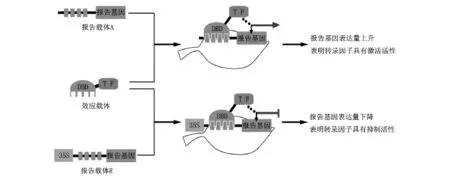
图1 转录因子调控活性的鉴定
35S启动子驱动的某些对植物早期生长发育起重要作用的转录因子会引起致死效应,这种情况下可采用基因诱导表达法,即将植物转录因子基因置于诱导型启动子下游,如酒精诱导启动子[31],目的基因只有在酒精诱导处理的条件下才会表达,正常条件下不对植物生长发育造成影响。也可将转录因子与激素受体系统相融合。如将植物转录因子与糖皮质激素受体(glucocorticoid receptor,GR)或雌激素受体(estrogen,ER)相融合[32-33],无配体结合时转录因子活性受到抑制,施与此类激素后,转录因子活性得以恢复。
基因功能增加表型分析已在鉴定基因功能上得到广泛应用,但它也有一定的局限性。很多转录因子是以蛋白复合体的形式行使功能的,只过量表达其中一个蛋白质,植物不会产生明显的表型变化。另外,转录因子的过量表达并不总是引起下游基因的变化,因为有些基因的表达是转录因子和其他因素共同作用的结果。
3.2 基因功能缺失表型分析
与基因功能增加相比,基因功能缺失的表型能更好地体现基因的生理功能。基因功能缺失突变体通常可通过插入法和反义RNA抑制法获得。插入法是在目的基因中插入DNA片段,使其无法正常表达,包括转座子插入法、T-DNA插入法和同源重组插入法。最常见的是T-DNA插入法,目前拟南芥人工突变体中有很多是通过该法获得的,这些突变体可通过拟南芥生物学资源中心(Arabidopsis Biologi⁃cal Resource Center,ABRC)或欧洲拟南芥种质中心(European Arabidopsis Stock Centre,NASC)查询并购买。T-DNA插入的局限性是基因编码区序列太短时,T-DNA插入的几率较小,插入非编码区或内含子区域时不一定会引起基因表达的完全缺失,只是使基因表达水平降低。反义RNA或RNA干扰(RNAi)技术是指将基因全部或部分的反向序列转入植物,所表达的RNA与目的mRNA相配对形成双链mRNA,从而导致mRNA的降解或抑制翻译。
人工 miRNA(artificial microRNA,amiRNA)是近年兴起的特异性引发基因沉默的有效方法[34-35],可用于抑制一个或几个相关基因的表达。与传统的RNAi相比,amiRNA来源于一个短的茎环结构,只产生一个单一的小RNA,能更加精准地抑制靶基因的表达。目前已有软件Web MicroRNA designer(http://weigelword.org)可用于amiRNA序列的设计[34]。
利用基因缺失来研究转录因子功能的最大挑战是,某些同一家族的转录因子因具有相似的DNA结合域,可调控相同的下游基因的表达,产生功能的冗余,单一基因的敲除很难产生明显表型。嵌合抑制基因沉默技术(chimeric repressor gene-silencing technology,CRES-T)[36]可解决这一难题。CRES-T是在转录因子基因上融合一个经过修饰的抑制结构域,并使其过量表达。这个嵌合的转录因子结合到靶基因启动子上可以有效地抑制基因的转录,并且使结合到该启动子上的其他转录因子的激活作用失效,这相当于敲除了结合到同一启动子区域的所有转录因子,得到明显表型的几率大大增加。当然,对于抑制基因表达的植物转录因子,CRES-T技术是不适用的,抑制结构域的嵌合对于具有抑制作用的转录因子实际上起到了基因功能增加的效果。
4 调控网络和组学分析
转录因子行使生理功能的过程十分复杂,涉及到所调控的靶基因、相关下游基因及与其他转录因子的相互作用。转录调控网络的建立和组学研究的应用,能使转录因子研究更加深入和系统。
4.1 调控网络分析
在转录因子及其靶基因和相关的下游基因之间建立相互联系的网络系统,能更全面清楚地了解转录因子的功能。传统的遗传学方法通过基因过表达或缺失突变体的异常表型来分析转录因子的功能,能够使这些异常表型得以全部或部分恢复的基因就是与该转录因子密切相关的下游基因。如拟南芥FUL基因编码MADS-box转录因子,ful突变体会表现出充满种子的短角果性状,如果将这个突变体中的 INDEHISCENT(IND)、SHATTERPROOF1(SHP1)和SHP2基因敲除,突变体的异常表型会在很大程度上得以修复。该修复现象表明ful突变体中IND、SHP1和SHP2基因的表达量升高,说明IND、SHP1和SHP2基因是FUL转录因子调控的下游基因,且FUL对三者的表达具有抑制调控作用[37]。
酵母单杂交是一种高通量鉴定转录因子与靶基因之间关系的方法。根据转录因子与顺式作用元件结合调控报告基因表达的原理,以一系列待研究的启动子中的顺式作用元件为诱饵,筛选含有转录因子的cDNA文库[38]。传统酵母单杂交方法耗时费力,且从启动子序列中确定顺式作用元件十分复杂。Deplancke等对此法进行了改良,以启动子中500 bp区域替代顺式作用元件为诱饵,筛选只包含转录因子的cDNA文库,效率大大提高[39]。如拟南芥中CCA1 HIKING EXPEDITION/TCP21可直接调控CIRCADIAN CLOCK ASSOCIATED 1(CCA1)的表达就是通过该方法鉴定出来的[40]。这种改良的酵母单杂交方法在转录因子调控网络研究中潜力巨大。
染色体免疫沉淀(chromatin immunoprecipita⁃tion,ChIP)是研究体内蛋白质与DNA相互作用的有效方法,可用于转录因子靶基因的筛选。ChIP是在活细胞状态下用甲醛固定蛋白质-DNA复合物,然后用酶切或超声波把染色体分离并打碎为一定大小的片段,利用特异性的抗体结合染色体片段上转录因子,沉淀蛋白质-DNA复合物,通过水解释放复合体中DNA,得到目的DNA片段的序列信息。ChIP与基因芯片相结合的ChIP-chip技术应用范围更加广泛,可用于转录因子靶基因的高通量筛选。将ChIP获得的DNA序列与基因芯片杂交,可实现基因组范围内的靶基因启动子的筛选,如果有可能获得专门应用于ChIP-Chip技术中的启动子芯片,则筛选的效率更高[41-42]。
此外,还有一种嵌合转录因子技术也可用于靶基因的鉴定。在转录因子上融合一个激活域,如VP16-AD和一个诱导受体,如GR或ER,将融合基因转入植物,用相应的诱导激素处理转基因植株,转录因子便会表现活性,促进靶基因的表达,同时与靶基因密切相关的下游基因的表达也会发生变化。用基因芯片技术对比分析激素处理前后基因转录水平,可以鉴定出靶基因和相关的下游基因,但这种情况下无法将靶基因和相关的下游基因区分开来。如果用诱导激素和环己酰亚胺(cycloheximide,CHX,一种蛋白质合成抑制剂)同时处理,则转录因子被激活,促进靶基因的表达,由于CHX的作用,新的蛋白质无法合成,新形成的靶基因mRNA无法翻译成蛋白质,由其引起的相关下游基因的表达不会受到影响[43]。用基因芯片技术对比分析处理前后基因的转录水平,即可获得转录因子直接作用的靶基因的相关信息。
对模式植物拟南芥来说,还可以通过查询相关数据库,如 AGRIS(Arabidopsis Gene Regulatory In⁃formation Server,http://arabidopsis.med.ohio-state.edu/)了解基因转录调控的相关信息。AGRIS数据库包含拟南芥转录因子数据库(AtTFDB)、拟南芥顺式作用元件数据库(AtcisDB)和拟南芥调控网络数据库(AtRegNet)。其中AtTFDB和AtcisDB主要提供转录因子及其启动子的相关信息,而AtRegNet能分析转录因子与其靶基因的调控关系,并可绘制网络图谱。此外,AtRegNet还开发了ReIN(Regulatory Networks Interaction Module)工具软件(http://arabi⁃dopsis.med.ohio-state.edu/REIN),这个软件可将上述3个数据库的资料加以整合,并可根据用户的要求计算并建立扩展的基因调控网络[44]。
4.2 组学分析
组学研究内容庞大,包括基因组学、转录组学、蛋白组学、代谢组学和表型组学等。在植物中,转录水平的调控对基因表达起重要作用,有些转录因子是多种重要生理活动的关键调控因子,其基因突变会引起靶基因以外的调控网络中大量基因的改变。通过转录组、蛋白质组、代谢组及表型组的研究,可更全面地揭示转录因子的生理功能。进一步将CRES-T、ChIP、基因芯片等技术应用于组学研究,为转录因子功能分析提供了新的途径。
[1]Paz-Ares J,Ghosal D,Wienand U,et al.The regulatory c1 locus of Zea mays encodes a protein with homology to myb proto-oncogene products and with structuralsimilarities to transcriptional activators[J].EMBO J,1987,6(12):3553-3558.
[2]Riechmann J L,Heard J,Martin G,et al.Arabidopsis tran⁃scription factors:genome-wide comparative analysis among eu⁃karyotes[J].Science,2000,290(5499):2105-2110.
[3]Finkelstein R R,Lynch T J.The Arabidopsis abscisic acid re⁃sponse gene ABI5 encodes a basic leucine zipper transcrip⁃tion factor[J].Plant Cell,2000,12(4):599-610.
[4]张全琪,朱家红,倪燕妹,等.植物bHLH转录因子的结构特点及其生物学功能[J].热带亚热带植物学报,2011,19(1):84-90.
[5]Singh K,Foley R C,Oñate-Sánchez L.Transcription factors in plant defense and stress responses[J].Curr Opin Plant Bi⁃ol,2002,5(5):430-436.
[6]才华,朱延明,柏锡,等.野生大豆GsbZIP33基因的分离胁迫耐性分析[J].分子植物育种,2011,9(4):397-401.
[7]Sønderby I E,Burow M,Rowe H C,et al.A complex inter⁃play of three R2R3 MYB transcription factors determines the profile of aliphatic glucosinolates in Arabidopsis[J].Plant Physiol,2010,153(1):348-363.
[8]李宗艳,李名扬.调控植物类黄酮生物合成的转录因子研究进展[J].南京林业大学学报:自然科学版,2011,35(5):129-134.
[9]Matys V,Fricke E,Geffers R,et al.TRANSFAC:transcrip⁃tionalregulation,from patterns to profiles[J].Nucleic Acids Res,2003,31(1):374-378.
[10]Riaño-Pachón D M,Ruzicic S,Dreyer I,et al.PlnTFDB:an integrative plant transcription factor database[J].BMC Bioinfor⁃matics,2007,8:42.
[11]Pérez-Rodríguez P,Riaño-Pachón D M,Corrêa L G,et al.PlnTFDB:updated content and new features of the plant tran⁃scription factor database[J]. Nucleic Acids Res, 2010,38:D822-D827.
[12]Mitsuda N,Ohme-Takagi M.Functional analysis of transcrip⁃tion factors in Arabidopsis[J].Plant Cell Physiol,2009,50(7):1232-1248.
[13]Gao G,Zhong Y,Guo A,et al.DRTF:a database of rice transcription factors[J].Bioinformatics,2006,22(10):1286-1287.
[14]Simon M,Lee M M,Lin Y,et al.Distinct and overlapping roles of single-repeat MYB genes in root epidermal patterning[J].Dev Biol,2007,311(2):566-578.
[15]Kagaya Y,Ohmiya K,Hattori T.RAV1,a novel DNA-bind⁃ing protein,binds to bipartite recognition sequence through two distinct DNA-binding domains uniquely found in higher plants[J].Nucleic Acids Res,1999,27(2):470-478.
[16]Quevillon E,Silventoinen V,Pillai S,et al.InterProScan:pro⁃tein domains identifier[J].Nucleic Acids Res,2005,33:W116-W120.
[17]Bailey T L,Williams N,Misleh C,et al.MEME:discovering and analyzing DNA and protein sequence motifs[J].Nucleic Acids Res,2006,34:W369-W373.
[18]Heazlewood JL,Verboom R E,Tonti-FilippiniJ,etal.SUBA:the Arabidopsis subcellular database[J].Nucleic Acids Res,2007,35:D213-D218.
[19]Emanuelsson O,Nielsen H,Brunak S,et al.Predicting subcel⁃lular localization of proteins based on their N-terminal amino acid sequence[J].J Mol Biol,2000,300(4):1005-1016.
[20]Hua S,Sun Z.Support vector machine approach for protein subcellular localization prediction[J].Bioinformatics,2001,17(8):721-728.
[21]Horton P,Park K J,Obayashi T,et al.WoLF PSORT:protein localization predictor[J].Nucleic AcidsRes,2007,35:W585-W587.
[22]Larue C T,Wen J,Walker J C.A microRNA-transcription factor module regulates lateral organ size and patterning in Arabidopsis[J].Plant J,2009,58(3):450-463.
[23]Zimmermann I M,Heim M A,Weisshaar B,et al.Comprehen⁃sive identification of Arabidopsis thaliana MYB transcription factors interacting with R/B-like BHLH proteins[J].Plant J,2004,40(1):22-34.
[24]Ohta M,Matsui K,Hiratsu K,et al.Repression domains of class II ERF transcriptional repressors share an essential mo⁃tif for active repression[J].Plant Cell,2001,13(8):1959-1968.
[25]黄泽军,黄荣峰,黄大昉.植物转录因子功能分析方法[J].农业生物技术学报,2002,10(3):295-300.
[26]Ueki S,Lacroix B Krichevsky A,et al.Functional transient genetic transformation of Arabidopsis leaves by biolistic bom⁃bardment[J].Nat Protoc,2009,4(1):71-77.
[27]Ichikawa T,Nakazawa M,Kawashima M,et al.The FOX hunting system:an alternative gain-of-function gene hunting technique[J].Plant J,2006,48(6):974-985.
[28]耿微,高野哲夫,柳参奎.FOX Hunting System—一种新的基因功能筛选技术[J].分子植物学育种,2010,8(4):810-817.
[29]Higuchi M,Kondou Y,Ichikawa T,et al.Full-length cDNA overexpressorgene hunting system(FOX hunting system)[J].Methods Mol Biol,2011,678:77-89.
[30]Fujita M,Mizukado S,Fujita Y,et al.Identification of stress⁃tolerance-related transcription-factor genes via mini-scale full-length cDNA over-expressor(FOX)gene hunting system[J].Biochem Biophys Res Commun,2007,364(2):250-257.
[31]Peebles C A,Gibson S I,Shanks J V,et al.Characterization of an ethanol-inducible promoter system in Catharanthus roseus hairy roots[J].Biotechnol Prog,2007,23(5):1258-1260.
[32]Aoyama T,Chua N H.A glucocorticoid-mediated transcription⁃al induction system in transgenic plants[J].Plant J,1997,11(3):605-612.
[33]Zuo J,Niu Q W,Chua N H.Technical advance:An estrogen receptor-based transactivator XVE mediates highly inducible gene expression in transgenic plants[J].Plant J,2000,24(2):265-273.
[34]Schwab R,Ossowski S,Riester M,et al.Highly specific gene silencing by artificial microRNAs in Arabidopsis[J].Plant Cell,2006,18(5):1121-1133.
[35]Niu Q W,Lin S S,Reyes J L,et al.Expression of artificial microRNAs in transgenic Arabidopsis thaliana confers virus re⁃sistance[J].Nat Biotechnol,2006,24(11):1420-1428.
[36]Hiratsu K,Matsui K,Koyama T,et al.Dominant repression of target genes by chimeric repressors that include the EAR motif,a repression domain,in Arabidopsis[J].Plant J,2003,34(5):733-739.
[37]Liljegren S J,Roeder A H,Kempin S A,et al.Control of fruitpatterning in Arabidopsis by INDEHISCENT[J].Cell,2004,116(6):843-853.
[38]陈峰,李洁,张贵友,等.酵母单杂交的原理与应用实例[J].生物工程进展,2001,21(4):57-62.
[39]Deplancke B,Mukhopadhyay A,Ao W,et al.A gene-cen⁃tered C.elegans protein-DNA interaction network[J].Cell,2006,125(6):1193-1205.
[40]Pruneda-Paz J L,Breton G,Para A,et al.A functional ge⁃nomics approach reveals CHE as a component of the Arabi⁃dopsis circadian clock[J].Science,2009,323(5920):1481-1485.
[41]张娜,尹继刚,孙高超,等.真核生物转录因子及其研究方法进展[J].动物医学进展,2009,30(1):75-79.
[42]李敏俐,王薇,陆祖宏.ChIP技术及其在基因组水平上分析DNA与蛋白质相互作用[J].遗传,2010,32(3):219-228.
[43]Samach A,Onouchi H,Gold S E,et al.Distinct roles of CONSTANS target genes in reproductive development of Ara⁃bidopsis[J].Science,2000,288(5471):1613-1616.
[44]Yilmaz A,Mejia-Guerra M K,Kurz K,et al.AGRIS:the Arabidopsis gene regulatory information server,an update[J].Nucleic Acids Res,2011,39:D1118-D1122.
