共轭烃中的非键分子轨道
2013-08-16刘柏平
刘 振,刘柏平
(华东理工大学化工学院,化学工程联合国家重点实验室,上海200237)
休克尔分子轨道理论的假设有3点[1-2]:(1)σ键和π键在分子中互不作用;(2)不相临原子间的轨道重叠为零;(3)不相临原子间的相互作用能为零.根据这个假设,各个原子中的内层电子以及形成σ键的电子均出现在局域化的轨道中,而离域的π电子则可以自由地在分子骨架内出现.因此,分子的诸多性质包括共振能,键级的变化以及电荷密度的分布都由自由的π电子决定[3].利用休克尔分子轨道理论,可以使用相对简单的数学处理方法,对诸多平面共轭小分子体系的性质进行定性的描述.即便是在从头算计算化学盛行的今天,基于自洽场迭代求解的HF轨道初始值,依然由扩展的休克尔分子轨道方法来求得.对于一些特殊的共轭交替烃分子,由于在休克尔分子轨道矩阵的特征值中存在一个零值,这个特征值为零的分子轨道便被称为非键分子轨道(Non-Bonding Molecular Orbital,NBMO).对于含有非键分子轨道的交替共轭烃分子体系,通过简单的“星号标注法”来求解其NBMO的表达式,以便快速地对该类分子的一些化学反应特性进行判断.
交替共轭烯烃是一类重要的含有非键分子轨道的分子体系,最早由COULSON和LONGUET-HIGGINS[3-4]在研究共轭分子体系电子密度、键级、相互极化率时作为特例提出.交替共轭,也就是说如果将碳骨架用星号(*)交替标记,则分子中不会出现2个星号碳或者非星号碳相邻的现象(图1).

图1 交替共轭分子和非交替共轭分子示例Figure 1 Alternant conjugated hydrocarbons and non-alternant conjugated hydrocarbons
根据分子中碳骨架原子数目的不同,交替共轭分子又分为奇数碳交替共轭化合物和偶数碳交替共轭化合物.由于该类分子中存在非键分子轨道,根据简单的“星号标注法”就可以快速求得此类分子体系的非键分子轨道和各个碳原子上的电荷密度,判断该类分子的稳定性及其化学反应性质[5-7].国内外对非键分子轨道的研究也有诸多报道[7-15].本文结合“星号标注法”求解奇、偶交替共轭烯烃分子中非键分子轨道的实例,结合DFT计算,对交替共轭烃中的非键分子轨道进行了讨论与分析.
1 非键分子轨道计算方法
1.1 奇数碳交替共轭烃的非键分子轨道
对于一些特殊的有机分子体系,比如含有奇数个π电子中心的交替共轭有机分子,由于其分子轨道中存在一个非键的HOMO(最高被占轨道),LONGUET-HIGGINS[5]提出,可以采用简便的“星号标注法”,快速求得非键分子轨道(NBMO)的表达式.下面将以苄基自由基分子(图2A)为例,介绍“星号标注法”求解非键分子轨道的基本步骤.
(1)首先对分子中的碳骨架进行序号标注,并将碳原子交替标注星号,且保证标注星号的数目最大(图2B).
(2)将不同星号碳原子的系数用a的正负整数倍表示,使得与非星号碳原子直接相连的所有星号碳原子的系数加和为零(即“零和规则”)(图2C).
(3)将所有碳原子系数进行归一化,并求出a的值.其中非星号碳原子系数为零.对于图2C中所示的分子,其NBMO的系数满足


式中,χi(i=1,3,5,7)为第 i个碳原子上的原子轨道.

图2 苄基自由基的分子结构及星号标注Figure 2 Schematic representation of benzyl radical and the star notation of the active carbons
1.2 偶数碳交替共轭烃的非键分子轨道
当采用“星号标注法”处理某些含有偶数碳原子的交替共轭烃分子时,必须将其剖析成多个具有非键分子轨道的奇数碳交替共轭烃,同时会出现偶数个非键分子轨道.
曹玲华等[8]对6种偶碳交替共轭烃中的非键分子轨道进行了研究(图3).在此对文献[8]中(Ⅴ)和(Ⅵ)进行了补充和完善.

图3 偶碳交替共轭烃Figure 3 Even alternant conjugated hydrocarbons
对于图3中,分子(I)的NBMO系数满足
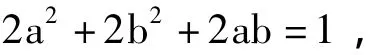
显然,
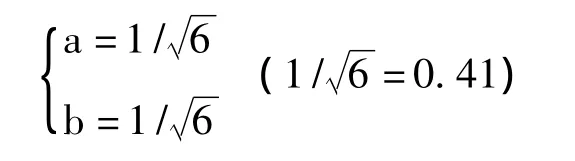
和
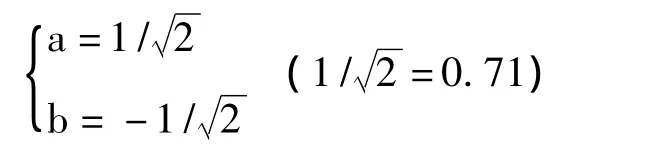
满足上式,故

同样,对于(Ⅱ)-(Ⅴ)4个分子,由于它们仍然只有2个非键分子轨道,因此应该设定a、b等2个变量.而分子(Ⅵ)的NBMO的个数为4(n-2×npi=14-2×5=4),所以该分子具有4个非键轨道,需设定a、b、c、d等4个变量.这5种分子的非键分子轨道的原子轨道系数标注见图4,其满足的条件关系式为:
(Ⅱ)3a2+3b2+4ab=1,
(Ⅲ)7a2+6ab+3b2=1,
(Ⅳ)17a2+28ab+17b2=1,
(Ⅴ)22a2+66ab+66b2=1,
(Ⅵ)5a2+5b2+3c2+3d2+6ab-2ad-2bc=1.
因此,从上述方程的解,可以求得非键分子轨道的原子轨道系数,构建各分子的非键分子轨道.
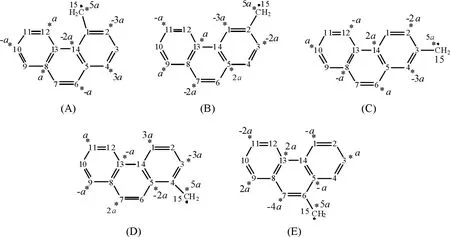
图4 菲亚甲基自由基的分子结构及星号标注Figure 4 Schematic representations of5 kinds ofmethylene-phenanthrene radicals and the star notation of the active carbons
1.3 DFT 计算说明
为了验证“星号标注法”的计算结果,作者基于Gaussian09软件包,采用B3LYP泛函对本文中讨论的几种典型的有机分子的非键分子轨道进行了DFT计算.在此,需要得到的信息是各化合物的非键分子轨道组成及各碳原子处的电荷密度分布,并不需要求得该分子的精确能量.为了便于分子轨道组成的分析,在DFT计算过程中采用了sto-3g基组函数.在非键分子轨道分析中,采用了原子轨道线性组合成分子轨道(LCAO-MO)的方法对DFT计算结果进行分析.
2 结果与讨论
2.1 奇数碳交替共轭烃的非键分子轨道
从非键分子轨道的各原子轨道系数,可以求得各个碳原子上的电荷密度.表1列出了根据“星号标注法”求得的苄基自由基分子各碳原子处的电荷密度.可以看出,C2、C4和C6原子处的系数均为零,说明该类原子对NBMO的贡献为零.而NBMO中的π电子则分布于整个分子骨架中标号为奇数的碳原子上.在亚甲基处的电荷密度最大,比较容易发生亲电取代反应,其余1、3、5位置的碳原子的电荷密度仅为C7处电荷密度的1/3左右,反应活性次之,而2、4、6位置则不易发生化学反应.

表1 “星号标注法”得到的苄基自由基非键分子轨道组成及其相对电荷密度分布Table 1 The coefficients and electron densities of the NBMO of benzyl radical calculated by“star notation”method
基于LCAO-MO轨道分析方法,将苄基自由基的非键分子轨道的DFT计算结果列入表2中.通过对比表1与表2的数据,发现采用简单的“星号标注法”计算得到的苄基自由基非键分子轨道的原子轨道的线性组合系数,以及电荷密度分布的结果与DFT的计算结果总体上是一致的.即亚甲基中的C7原子中的电荷密度最大,也说明非键分子轨道中的自由π电子在C7原子处出现的几率最高,因此亚甲基也具有最高的反应活性.其次,在苯环中的6个碳原子,根据“星号标注法”得到的C1、C3和C5中的电荷密度相等,具有相同的反应活性,而C2、C4和C6中的电荷密度为零.这是因为休克尔分子轨道理论不考虑相邻原子间的相互作用,并且忽略了分子中可能存在的极化作用,从而使得基于休克尔分子轨道理论的“星号标注法”的计算结果中,同一类碳原子上的电荷密度是均匀分配的.而DFT的计算结果表明,C2、C4和C6中虽然也有电子出现,但电荷密度极低,几乎忽略不计,而位于对称轴上的C3原子处的电荷密度为0.202,略微高于C1和C5处的电荷密度0.16.以上数据表明,“星号标注法”完全适用于快速确定奇数碳交替共轭平面有机小分子的反应活性.

表2 DFT计算得到的苄基自由基非键分子轨道组成及其相对电荷密度分布Table 2 The coefficients and electron densities of the NBMO of benzyl radical calculated by DFT
对于该类自由基分子体系,其非键分子轨道中含有1个未成对的自由π电子.当将π电子从非键分子轨道中移除形成阳离子,或者从外部获得1个电子形成阴离子时,仅会影响非键分子轨道中各原子轨道的电荷密度大小,不会改变非键分子轨道的形状和组成.作者通过DFT计算,将得到的苄基自由基、苄基阳离子和苄基阴离子的非键分子轨道的电荷密度以及非键分子轨道图形一并在表3中给出.苄基自由基、苄基阳离子和苄基阴离子的非键分子轨道的几何形状是等同的,没有因电子的得失而反生变化,其主要的贡献来自于C1、C3、C5和C7的p轨道.从轨道示意图可以看出,C1和C5原子轨道之间发生了部分重叠,并包含了部分来自C6原子p轨道的贡献.而“星号标注法”并不考虑这种相互作用,因而其得到的C6原子处的电荷密度为零.表3中的数据表明,当苄基自由基得到1个电子形成苄基阴离子时,由于非键分子轨道中排布了2个电子,从而在各奇数标号原子处的电荷密度比苄基自由基增加了1倍,而相对比例并没有发生变化.至于将非键分子轨道中的电子移除后得到的苄基阳离子,由于非键分子轨道中没有电子出现,从而各原子处的电荷密度也为零.

表3 DFT计算得到的苄基自由基、苄基阳离子和苄基阴离子的非键分子轨道及电荷密度Table 3 The electron densities of the NBMO of benzyl radical,benzyl cation,and benzylanion,calculated by DFT.Also shown is the graphical representation of the NBMO of the benzyl radical
以上针对简单的苄基自由基分子体系,介绍了采用“星号标注法”确定其非键分子轨道的具体步骤,并与DFT的计算结果进行了对比分析.对于略微复杂的大分子体系,采用NBMO方法还可以快速比较当取代基位置不同时,其分子骨架中的电荷密度分布情况,用以判断各自由基的反应活性.下面再以5种不同的菲亚甲基自由基分子(图4)为例,进一步介绍“星号标注法”在确定一系列同分异构体的非键分子轨道中的应用.
作者仍采用之前介绍的“星号标注法”对图4中的5个菲亚甲基自由基进行标号,按照“零和规则”对各个碳原子进行标注.同样,将非键分子轨道的系数进行归一化,可以求得以上5种异构体的NBMO的表达式如下:
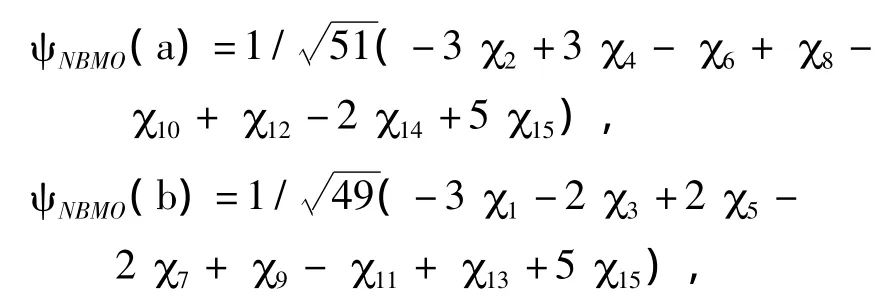
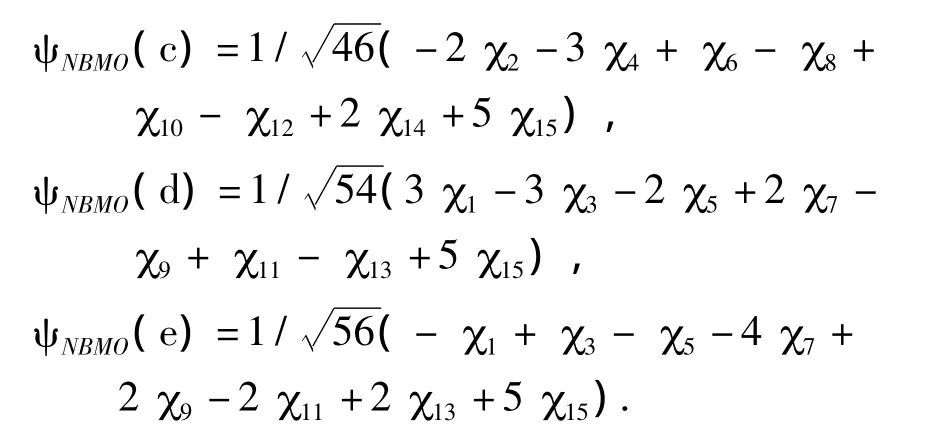
从非键分子轨道的各原子轨道系数,可以求得各个碳原子上的电荷密度(表4).对A结构而言,在1号碳为引入的亚甲基自由基基团破坏了菲分子的结构对称性,使得亚甲基所在的苯环中的电荷密度显著增加,其中2号碳同4号碳的电荷密度相等,且为稠环碳中最高的电荷密度,比较容易发生亲电取代反应,而其余位置的电荷密度则相对低.当亚甲基分别位于2、3、4、6位碳时,其各原子轨道系数的平方加和与A结构略微不同,其中E结构的系数平方和最大,而C结构的系数平方和最小.由于电荷密度与系数平方和成反比关系,因此,当亚甲基自由基位于菲分子稠环上3号碳(C结构)时,其电荷密度最大,具有最高的反应活性,且其邻位4号碳也容易发生亲电取代反应.当取代基位于6号碳(E结构)时,亚甲基自由基上电荷密度最低,但是其邻位碳在所有5种同分异构体的稠环碳中电荷密度最大,最容易发生亲电取代反应.
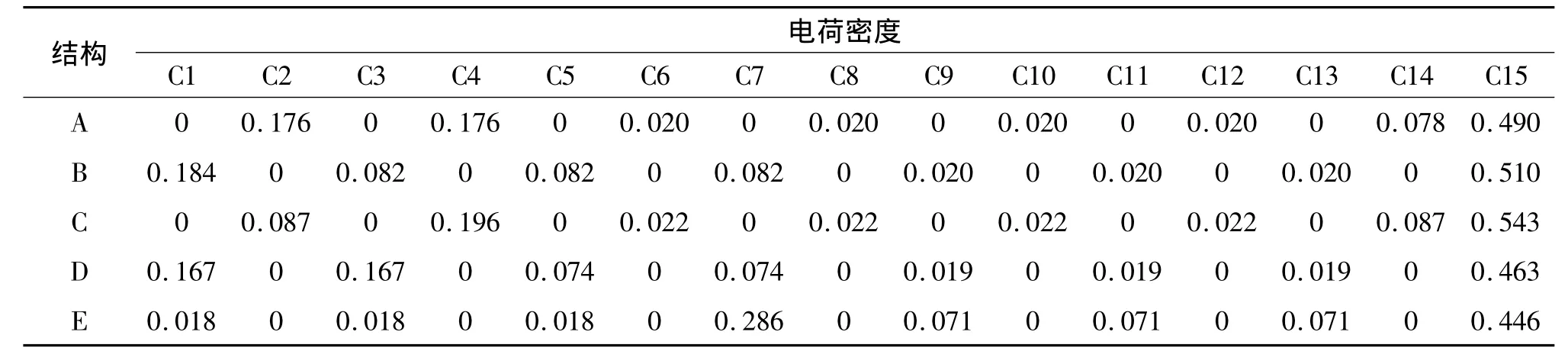
表4 菲亚甲基自由基(5种取代位置)的非键分子轨道的电荷密度分布Table 4 The electron densities of the NBMO of 5 kinds ofmethylene-phenanthrene radicals calculated by“star notation”method
通过“星号标注法”对上述5种分子的非键分子轨道的计算结果与DFT的计算结果基本是一致的(表4、表5).比如,“星号标注法”给出的亚甲基中C15上的电荷密度,除了结构B中的电荷密度略微偏高以外,其余4种结构中C15上的电荷密度与DFT的计算结果基本一致.另外一个比较明显的差别在于A结构中C12原子处的电荷密度,“星号标注法”给出的电荷密度为0.02,而DFT的计算结果则为0.008,这是因为在“星号标注法”中并没有考虑到A分子中亚甲基基团对C12原子的诱导效应,而实际分子构型中,A结构中的C15与C12之间存在着比较明显的排斥作用.尽管采用“星号标注法”得到的非键分子轨道的电荷密度分布与DFT的计算结果存在些许差异,但是在总体上还是一致的.
因此,对于奇数交替共轭烃,基于标注星号待定系数法,可以快速确定其非键分子轨道(NBMO)的电荷密度分布,并用于比较在不同位置引入取代基时,各位点碳上的亲电取代反应活性.

表5 菲亚甲基自由基(5种取代位置)的非键分子轨道的电荷密度分布Table 5 The electron densities of the NBMO of 5 kinds ofmethylene-phenanthrene radicals calculated by DFT
2.2 偶数碳交替共轭烃的非键分子轨道
通过DFT的计算结果,将图3中分子(I)的2个非键分子轨道以图形化的方式在图5中给出,轨道下方为NBMO的原子轨道线性组和公式.从1.2节中,知道根据“星号标注法”计算得到的三亚甲基甲烷自由基分子中NBMO(1)中C2、C3和C4的原子轨道系数分别为0.41、0.41 和 -0.82,而 NBMO(2)中 C2 和C3的原子轨道贡献分别为0.71和-0.71.这是由于在“星号标注法”中并没有考虑各个碳原子之间的相互作用,从而在非键分子轨道中C2和C3原子的贡献是等同的.而DFT的计算结果则表明,尽管在NBMO(1)中C4原子轨道的贡献是最大的,但是C2和C3原子在NBMO(1)和NBMO(2)中的贡献均略有不同.其中,C2的原子轨道在NBMO(1)中的贡献略微高于C3原子,而在NBMO(2)中则略微低于C3原子的轨道贡献.通过对比上述的计算结果可以发现,“星号标注法”在偶碳交替中也能够给出与DFT方法非常接近的计算结果.偶数碳交替共轭烃分子,其必定包含多个非键分子轨道,可以采用设置多变量的方法确定其系数.本文采用DFT计算,对含有非键分子轨道的几种典型分子进行了轨道分析,并与“星号标注法”的计算结果进行了对比分析.研究发现,由于“星号标注法”忽略了原子之间的相互作用,在计算所得的非键分子轨道中各原子轨道的贡献是平均的,而DFT计算则更能真实地反映分子中各原子之间的相互作用和分子内的极化作用.总体而言,“星号标注法”可以被快速用于确定交替烃分子中的非键分子轨道,并能够被用以判断分子内各碳原子处的反应活性.此外,由于“星号标注法”在确定非键分子轨道系数的过程中,对原子轨道系数进行了归一化处理,因此,在不同类型的分子间比较系数及电荷密度的大小不具有实际意义,该方法只能用来分析分子内各位点之间的反应活性大小,而不能用于对含有不同原子数目的分子之间比较.

图5 三亚甲基甲烷分子的非键分子轨道Figure 5 Graphical representations of the NBMOs of trimethylenemethane
3 结论
交替共轭烃分子中的非键分子轨道可以被快速确定.奇数碳交替共轭烃分子的NBMO系数比较容易采用“星号标注法”进行确定.而对于部分可拆分ings of the Royal Society of London:Series A:Mathematical and Physical Sciences,1947,192(1028):16-32.
[1]HÜCKEL E.Quantum-theoretical contributions to the benzene problem:I:The electron configuration of benzene and related compounds[J].Zeitschrift für Physik,1931,70(3/4):204-286.
[2]HÜCKEL E.Quantum theory treatment of the benzene problem:II:Quantum theory of induced polarity[J].Zeitschrift für Physik,1931,72(5/6):310-337.
[3]COULSON C A,LONGUET-HIGGINS H C.The Electronic structure of conjugated systems:I:General theory[J].Proceedings of the Royal Society of London:Series A:Mathematical and Physical Sciences,1947,191(1024):39-60.
[4]COULSON C A,LONGUET-HIGGINS H C.The Electronic structure of conjugated systems:II:Unsaturated hydrocarbons and their hetero-derivatives[J].Proceed-
[5]LONGUET-HIGGINSH C.Molecular-orbital theory:II:Ionization constants in heteroaromatic amines and related compounds[J].J Chem Phys,1950,18(3):275-282.
[6]DEWAR M J S.A molecular orbital theory of organic chemistry:VI:1 Aromatic Substitution and Addition[J].JAm Chem Soc,1952,74(13):3357-3363.
[7]LONGUET-HIGGINS H C.Molecular-orbital theory:I:Resonance structures and molecular orbitals in unsaturated hydrocarbons[J].JChem Phys,1950,18(3):265-274.
[8]曹玲华,张键.偶碳交替烃的非键分子轨道[J].新疆大学学报:自然科学版,1989,6(3):55-60.
[9]ITOH K.Electronic structures of aromatic hydrocarbons with high spin multiplicities in the electronic ground state[J].Pure Appl Chem,1978,50(11/12):1251-1259.
[10]BORDENW T,DAVIDSON E R.Effects of electron repulsion in conjugated hydrocarbon diradicals[J].J Am Chem Soc,1977,99(14):4587-4594.
[11]PRANATA J.Spin preferences of conjugated polyradicals:the disjoint NBMO analysis[J].JAm Chem Soc,1992,114(26):10537-10541.
[12]LANGLER R.How to recognize disjoint hydrocarbons:A Hückel theory topic[J].The Chemical Educator,2002,7(1):1-6.
[13]AOKIY,IMAMURA A.A simple rule to find nondisjoint NBMO degenerate systems for designing high-spin organic molecules[J].Int JQuantum Chem,1999,74(5):491-502.
[14]ORIMOTO Y,AOKIY.Analyticalmethod for predicting ferromagnetic properties of benzyl-radical polymers based on NBMO theory[J].JChem Theory Comput,2006,2(3):786-796.
[15]AOKI Y,IMAMURA A.A simple treatment to design NBMO degenerate systems in alternate and non-alternant hydrocarbons[J].Theoretica Chimica Acta,1992,84(3):155-180.
