新型4,6-脱水-α-D-吡喃型半乳糖衍生物的合成*
2013-03-26邵华武
柯 钧,邵华武
(1.中国科学院成都生物研究所天然产物研究中心,四川成都 610041;2.中国科学院大学,北京 100049)
D-半乳糖基寡糖及其缀合物在生物界广泛存在[1]。D-吡喃型半乳糖的6-位为活性较高的伯羟基,而4-位的仲羟基与其同位于糖环一侧,故在D-吡喃型半乳糖衍生物的合成中,通常用异丙亚基将 3-位,4-位[2]和苄亚基将 4-位,6-位同时保护[3],在不同反应体系下可选择性脱保护,从而控制反应的区域选择性。而4,6-脱水糖也可以同时实现4-位,6-位羟基保护,而四元环开环后还可以作为糖基受体参与糖苷化反应,但到目前为止这类化合物的研究报道较少。Goren等[4]报道了 2,3-二-O-苄基-6-对甲苯磺酰基-α-D-半乳糖在MeONa/MeOH体系中生成 4,6-脱水糖,收率66%;Magnusson 等[5]在 HMPT/CCl4,LiEt3BH/THF条件下制得4,6-脱水半乳糖,收率55%。
本文以甲基-2,3,4,6-四-O-苄基-α-D-吡喃型半乳糖苷(1)为原料,通过对1-位,2-位进行结构修饰,6-位选择性脱除苄基,再引入保护基Ms(或Ts)制得 3,4-二-O-苄基-6-磺酰基(或对甲苯基磺酰基)-α-D-吡喃型半乳糖衍生物(2a~2f);2在碱性条件下发生分子内亲核取代反应合成了一列新型的4,6-脱水-α-D-吡喃型半乳糖衍生物(3c~3f,Scheme 1),其结构经1H NMR,13C NMR 和ESI-HR-MS表征。并对反应条件进行了优化。合成3的最佳反应条件为,以乙酸钾/二甲亚砜为催化体系,于80℃反应24 h。
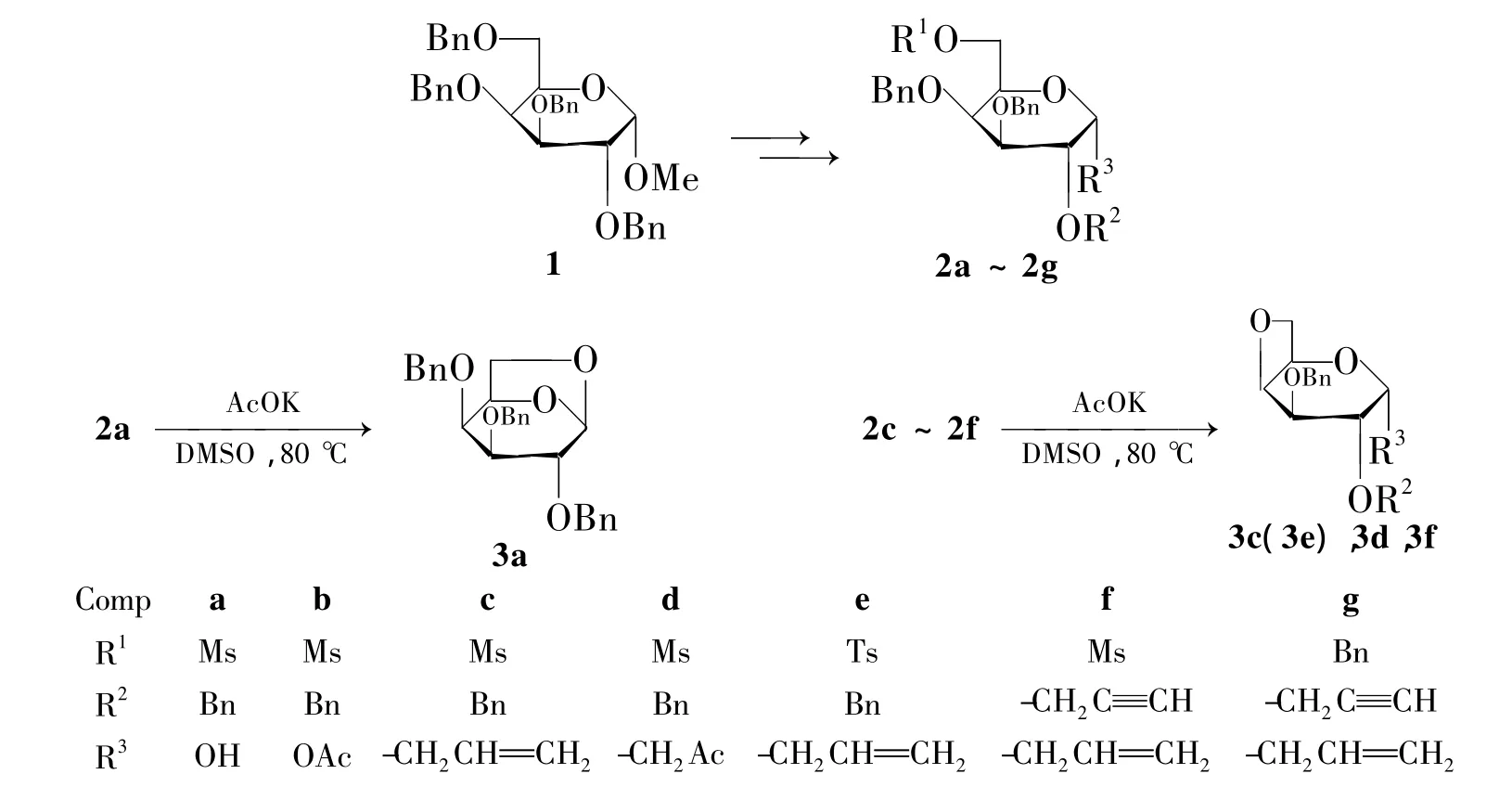
Scheme 1
1 实验部分
1.1 仪器与试剂
Avance Brucker-600 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);BioTOF Q型质谱仪;Perkin Elmer M341型自动旋光仪。
2a ~2e和2g 按文献[7,8]方法合成;硅胶,200目~400目,青岛海洋化工厂;其余所有试剂均为分析纯。
1.2 合成
(1)2f的合成[6]
将 2g[7,8]462 mg(0.90 mmol)溶于混合溶剂[V(AcOH)∶V(Ac2O)=1 ∶2,9 mL]中,搅拌下于0 ℃迅速加入无水 ZnCl21.23 g(9.0 mmol),滴毕,缓慢升至室温反应6 h。用混合溶剂A[V(乙酸乙酯)∶V(水)=1 ∶1,40 mL]萃取,合并有机层,依次用饱和NaHCO3溶液、饱和食盐水洗,无水Na2SO4干燥,浓缩后用MeOH(8 mL)溶解,加入K2CO3124 mg,于室温反应2 h。用混合溶剂A萃取,合并有机层,用经和食盐水洗涤,无水Na2SO4干燥,浓缩后用CH2Cl28 mL溶解。降温至0℃,依次滴加 Et3N 0.25 mL(1.80 mmol),MsCl 0.11 mL(1.35 mmol),滴毕,于室温反应6 h。用混合溶剂B[V(CH2Cl2)∶V(水)=1 ∶1,40 mL]萃取,合并有机层,用饱和食盐水洗涤,无水Na2SO4干燥,浓缩后经硅胶柱层析[洗脱剂:C=V(石油醚)∶V(乙酸乙酯)=6∶1]纯化得黄色浆状物2f 320 mg(3 步收率 71%),[α]20D+25.0°(c0.18);1H NMR δ:7.37 ~ 7.28(m,10H),5.80(m,1H),5.12(dd,J=17.2 Hz,1.5 Hz,1H),5.06(d,J=10.2 Hz,1H),4.97(dd,J=12.1 Hz,9.2 Hz,1H),4.74(d,J=12.0 Hz,1H),4.65(d,J=12.1 Hz,1H),4.61(d,J=11.8 Hz,1H),4.57(d,J=11.8 Hz,1H),4.43(dd,J=12.3 Hz,2.1 Hz,1H),4.26 ~ 4.22(m,1H),4.13(t,J=2.4 Hz,2H),4.07(t,J=6.3 Hz,1H),4.01(dd,J=5.7 Hz,2.8 Hz,1H),3.98 ~3.96(m,1H),3.63(d,J=4.4 Hz,1H),3.01(s,3H),2.39(dd,J=14.6 Hz,7.2 Hz,1H),2.36(t,J=2.2 Hz,1H),2.32(dd,J=13.7 Hz,6.7 Hz,1H);13C NMR δ:137.8,134.6,128.5,128.0,127.9,127.8,117.4,79.5,76.0,75.0,73.4,73.3,73.2,72.2,68.3,67.2,58.2,38.0,34.0;ESI-HR-MSm/z:Calcd for C27H32O7SNa{[M+Na]+}523.176 1,found 523.176 9。
(2)3的合成通法
将 2[6,7]1.50 mmol溶于 DMSO(2 mL)中,搅拌下加入AcOK 130 mg(4.07 mmol),于80℃反应至终点(TLC监测)。用混合溶剂A萃取,合并有机相,用饱和食盐水洗涤,无水Na2SO4干燥,浓缩后经快速硅胶柱层析(梯度洗脱剂:C=10∶1~6∶1)纯化得黄色浆状物3a,3c~3f。
3c:2c→3c,收率 83%;2e→3c(3e),收率78%,[α]20D+22.8°(c0.14);1H NMR δ:7.38 ~7.25(m,10H),5.75(m,1H),5.11(dd,J=17.2 Hz,1.2 Hz,1H),5.06(d,J=10.1 Hz,1H),4.54(s,2H),4.49(s,2H),4.42(d,J=5.3 Hz,1H),4.34(s,1H),4.28(s,1H),4.12(d,J=10.0 Hz,1H),4.03(dd,J=10.0 Hz,2.9 Hz,1H),3.86(m,1H),3.56(dd,J=5.3 Hz,2.8 Hz,1H),2.36(m,1H),2.33 ~ 2.27(m,1H);13C NMR δ:138.0,134.67,128.5,128.4,127.9,127.8,127.7,117.2,78.4,78.2,77.5,72.8,71.8,71.1,69.4,34.5;ESI-HR-MSm/z:Calcd for C23H26O4Na{[M+Na]+}389.172 3,found 389.171 4。
2 结果与讨论
以2c合成3c为模板,考察催化剂,溶剂,反应温度和反应时间对其收率的影响,结果见表1。从表1可见,碱性条件下该反应均能发生;在低极性非质子溶剂(CH2Cl2,THF)中该反应则未能发生(Entry 11,Entry 12),而在丙酮,MeCN 和MeOH中反应时间较长(Entry 13~Entry 15);同时反应需要的温度较高,在温度达到80℃时才能发生反应生成4,6-脱水糖,继续升高温度则对反应产率影响不大。
根据反应所得产物,我们推测其机理可能为4-O上的孤对电子进攻6-C,同时磺酰氧基在碱性条件下离去,然后4-苄基离去环化形成4,6-脱水糖。另外,1-C苷类底物反应产率较高;2a反应生成3a,可能是由于1-位的羟基构型转化后进攻6-C,新形成的五元环张力小于四元环[9];而1-位被乙酰基保护的2b在该条件下未能发生反应,则可能因为1-位羟基被吸电子基保护后导致分子内亲核性大大降低。
由此可见,合成3c的最佳反应条件为:以DMSO(2.0 mL)为溶剂,AcOK(1.0 eq)为碱,于温度80℃反应24 h,收率83%。
Goren[4]和 Magnusson 课题组[5]曾报道过 4,6脱水半乳糖合成方法,但存在收率低或操作复杂等缺点。而本文在AcOK/DMSO体系下实现了高收率地合成 4,6-脱水-α-D-吡喃型半乳糖,简便易行。由于在4,6-脱水糖中含有较大张力的四元环,在适当条件下可以开环,这也为半乳糖的4-位,6-位选择性糖苷化反应提供了一种新策略。
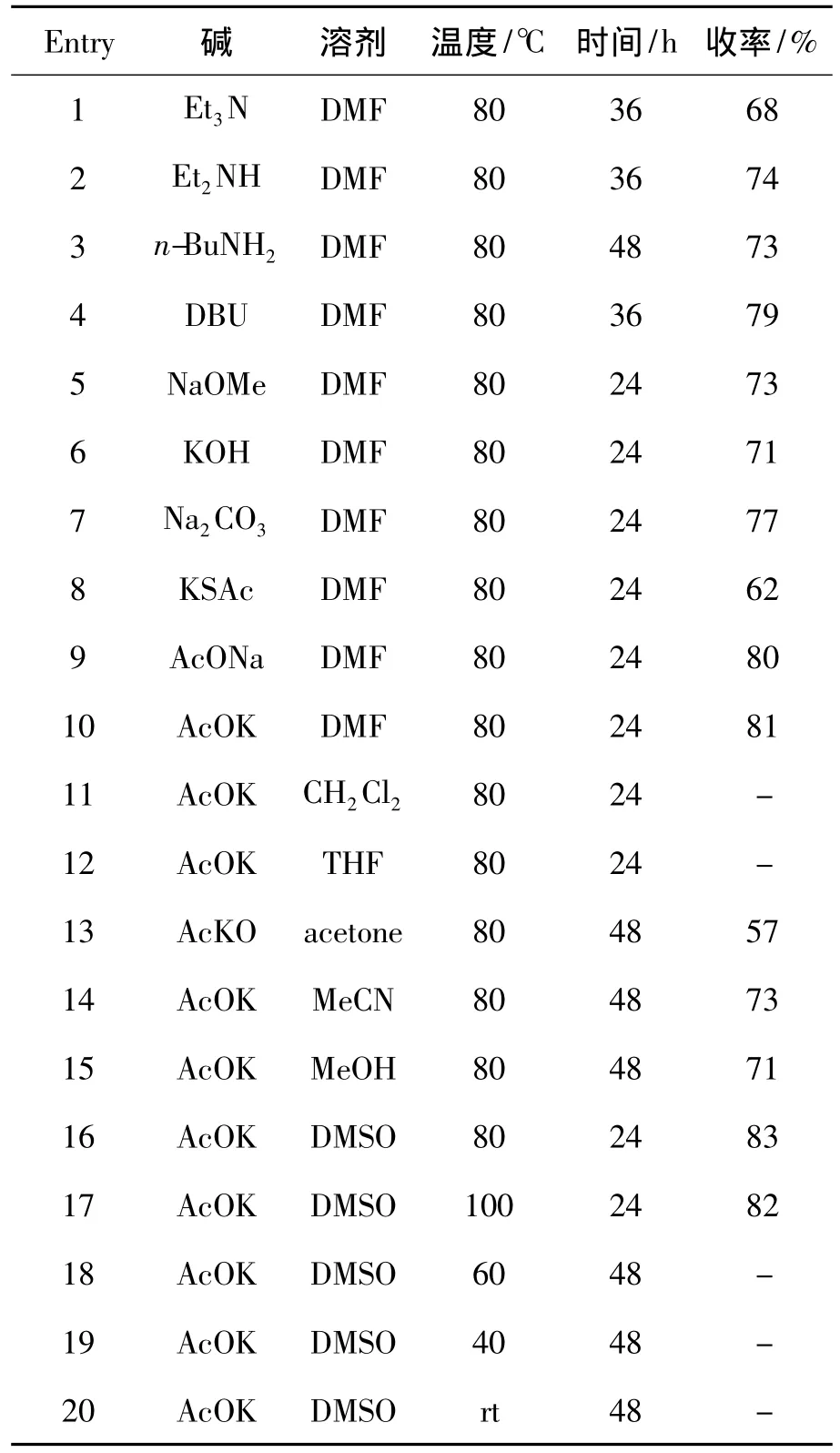
表1 反应条件对合成3c的影响*Table 1 Effect of reaction conditions on synthesizing 3c
[1]Miyagawa S,Takeishi S,Yamamoto A,et al.Survey of glycoantigens in cells from 1-3 galactosyl transferase knock out pig using a lectin microarray[J].Xenotransplantation,2010,17(1):61 -70.
[2]de Belder A N.Cyclic acetals of the aldoses and aldosides[J].Advances in Carbohydrate Chemistry,1965,20:219-302.
[3]Tanaka N,Ogawa I,Yoshigase S,et al.Regioselective ring opening of benzylidene acetal protecting group(s)of hexopyranoside derivatives by DIBAL-H[J].Carbohydrate Research,2008,(343):2675 -2679.
[4]Liav A,Goren M B,Yang Y,et al.Synthesis of 4,6-anhydro-α-D-galactopyranosyl-6-O-mycoloyl-and-corynomycoloyl-α-D-galactopyranoside[J].Carbohydrate Research,1986,(155):223 -228.
[5]Kihlberg J,Frejd T,Jansso K,et al.Synthetic receptor analogues:Prepration and calculated conformations of the 2-deoxy,6-O-methyl,6-deoxy,and 6-deoxy-6-fluoro derivatives of methyl 4-O-α-D-galactopyranosylβ-D-galactopyranoside(methyl β-D-galabioside)[J].Carbohydrate Research,1988,(176):271 -286.
[6]Yang G B,Ding X L,Kong F Z.Selective 6-O-debenzylation of mono-and disaccharide derivatives using ZnCl2-Ac2O-HAc[J].Tetrahedron Lett,1997,38:6725-6278.
[7]Shao H,Ekthawatchai S,Chen C S,et al.1,2-Migration of 2'-oxoalkyl group and concomitant synthesis of 2-C-branchedO-,S-glycosides and glycosyl azides via 1,2-cyclopropanated sugars[J].The Journal of Organic Chemistry,2005,70(12):4726 -4734.
[8]Cipolla L,Lay L,Nicotra F.New and easy access toC-glycosides of glucosamine and mannosamine[J].The Journal of Organic Chemistry,1997,62(19):6678 -6681.
[9]Lafont D,Boullanger P,Cadas O,et al.Mild procedure for the preparation of 1,6-anhydro-D-hexopyranoses and derivatives[J].Synthesis,1989:191 - 194.
