糖基化青蒿素衍生物的合成
2012-11-21任彦荣
任彦荣
(重庆第二师范学院 生物与化学工程系,重庆 400067)
青蒿素(Ⅰ)及其衍生物虽然在临床上应用多年,但仍存在油溶性和水溶性不佳、对热不稳定、易受湿、热和还原性物质的影响而分解、临床复发率高等缺点,使其应用受到限制[1~4]。糖类物质具有优良的性质,具有毒性小、结构新颖多样、靶向性好等特点。一些不溶于水的物质与糖结合转变成糖苷后,水溶解性增高,毒性降低。
本文通过化学方法将糖基引入Ⅰ分子中对其进行修饰,以期利用糖基的优良特性提高Ⅰ的水溶性,并降低毒性。以糖[D-葡萄糖(1a), D-木糖(1b), D-甘露糖(1c), 乳糖(1d)和D-半乳糖(1e)]为原料,经溴化、乙酰化一锅法使糖基乙酰化制得相应的溴代乙酰化糖(2a~2e); 2分别与二氢青蒿素(Ⅱ-H, Scheme 1)在相转移催化剂四丁基硫酸氢铵催化下经醚化反应合成了五个糖基化青蒿素衍生物(3a~3e, Scheme 2),其结构进行1H NMR,13C NMR, IR和LC-MS 表征。
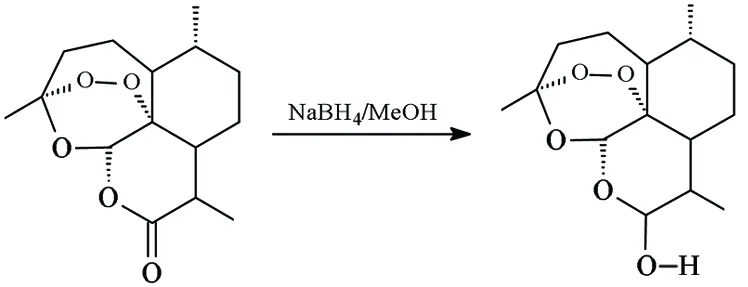
Ⅰ Ⅱ-H

Scheme2
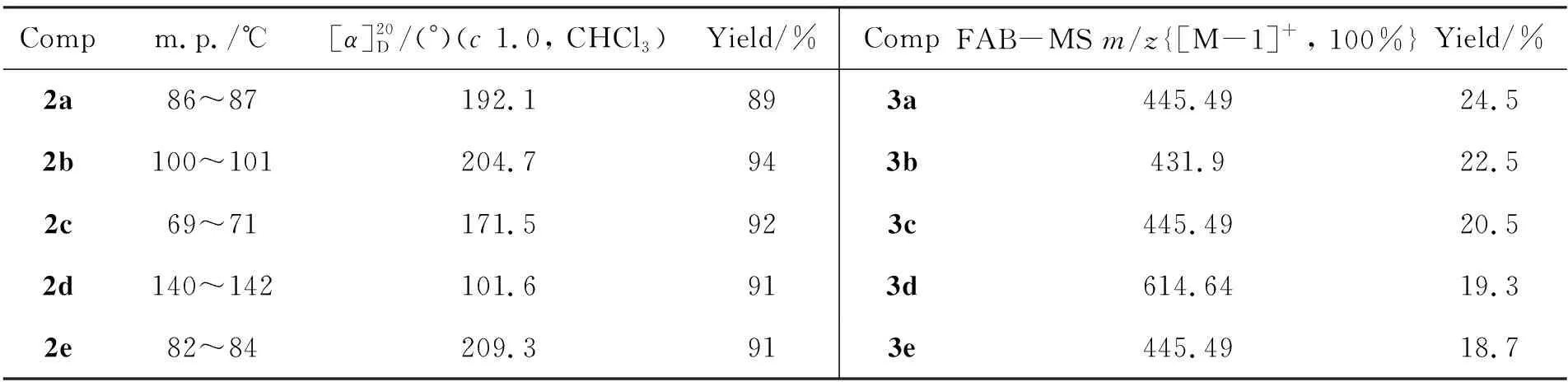
表1 合成2和3的实验结果Table 1 Experimental results of synthesizing 2 and 3
1 实验部分
1.1 仪器与试剂
Varian INOVA-400型核磁共振仪(CDCl3为溶剂,TMS为内标);Varon型傅立叶红外光谱仪(KBr压片);Finnigan-MAT 4510型质谱仪(EI)。
所用试剂均为分析纯,成都福瑞斯特科技发展有限公司。
1.2 合成
(1) Ⅱ-H的合成
在反应瓶中加入Ⅰ.2.81 g(10 mmol), NaBH40.454 g(12 mmol)及无水甲醇10 mL,搅拌下于室温反应24 h[TLC跟踪,展开剂:A=V(石油醚) ∶V(丙酮)=1 ∶1]。过滤,滤液减压蒸馏回收甲醇,残余物用250 mL水溶解,乙酸乙酯(3×50 mL)萃取,合并有机层,用少量水洗涤;合并水层与水洗液,再用乙酸乙酯萃取;合并有机层[5],蒸去溶剂,干燥得白色固体(±)-Ⅱ-H; IRν: 3 315, 2 935, 2 924, 1 134, 1 037, 1 047 cm-1。
(2) 2a~2e的合成通法[6]
在反应瓶中加入1a~1e2.00 g,醋酸酐悬浮液10 mL及碘90 mg,搅拌下于室温反应至反应液呈棕色透明(全乙酰化完成,5 min~30 min)。用二氯甲烷(50 mL)稀释,冰浴冷却下加入38%HBr/AcOH 14 mL,于室温反应至终点(TLC跟踪,展开剂A)。用二氯甲烷(150 mL)稀释后依次用冰水(2×60 mL),饱和碳酸氢钠溶液(2×50 mL)及0.4 mol·L-1硫代硫酸钠溶液(2×50 mL)洗涤;分液,有机层用无水硫酸钠干燥,减压浓缩得白色固体;经乙醚溶解后,用石油醚重结晶得白色针状晶体2a~2e(α-构型)。实验结果见表1,表征数据与文献[6]值一致。
(3) 3a~3e的合成通法
在反应瓶中加入二氯甲烷20 mL, 水15 mL及四丁基硫酸氢铵1.13 g,搅拌下于45 ℃(浴温)反应30 min;于1 h内同时滴加Ⅱ-H 180 g(4 mmol)的2.5%KOH(40 mL)溶液和2 4.8 mmol的二氯甲烷(20 mL)溶液。控制pH 9~10,继续反应16 h。用1 mol·L-1硫酸中和至中性,加入乙酸乙酯40 mL,减压抽滤,滤液分液,水相用乙酸乙酯(3×10 mL)萃取,合并有机相,用饱和氯化钠溶液(3×30 mL)洗涤,无水硫酸钠干燥12 h;抽滤,滤液减压浓缩得棕红色膏状物。将其溶于0.1 mol·L-1MeONa/MeOH(300 mL)中,于40 ℃反应24 h;过滤,滤饼用水50 mL溶解,蒸馏浓缩得固体,经柱色谱[洗脱剂:V(石油醚) ∶V(丙酮)=1 ∶1]纯化得白色固体3a~3e。实验结果和FAB-MS数据见表1。
3a:1H NMRδ: 5.43(s, 1H, CH), 4.90(d,J=6.2 Hz, 1H, CH), 4.76(d,J=3.2 Hz, 1H, CH), 3.32~3.58(m, 6H, CH, CH2,糖环-H), 1.28(s, 3H, CH3), 0.92(d,J=6.1 Hz, 3H, CH3), 0.83(d,J=6.0 Hz, 3H, CH3);13C NMRδ: 8.6, 13.9, 17.4, 18.2, 19.3, 23.7, 27.4, 27.7, 29.3, 30.5, 30.7, 62.5, 67.8, 69.0, 71.1, 72.1, 84.0, 92.4, 92.5, 95.1, 98.6; IRν: 3 379, 2 946, 2 924, 1 134, 1 027, 1 092 cm-1。
3b:1H NMRδ: 5.43(s, 1H, CH), 4.69(d,J=6.5 Hz, 1H, CH), 4.83(d,J=3.1 Hz, 1H, CH), 3.32~3.48(m, 5H, CH, CH2,糖环-H), 1.28(s, 3H, CH3), 0.91(d,J=6.2 Hz, 3H, CH3), 0.84(d,J=6.1 Hz, 3H, CH3);13C NMRδ: 8.6, 13.9, 18.5,21.8, 22.7, 23.6, 24.3, 25.0, 27.4, 27.5, 36.1, 65.3, 68.7, 71.2, 71.8, 86.8, 92.3, 92.4, 93.3, 94.7; IRν: 3 379, 2 946, 2 924, 1 134, 1 027, 1 041 cm-1。
3c:1H NMRδ: 5.44(s, 1H, CH), 5.01(d,J=1.5 Hz, 1H, CH), 4.72(d,J=3.7 Hz, 1H, CH), 3.24~3.68(m, 6H, CH, CH2,糖环-H), 1.28(s, 3H, CH3), 0.90(d,J=6.3 Hz, 3H, CH3), 0.83(d,J=7.2 Hz, 3H, CH3);13C NMRδ: 8.8, 14.5, 17.3, 18.7, 19.5, 24.0, 27.2, 29.9, 30.8, 30.5, 30.7, 62.5, 68.1, 69.0, 72.1, 84.3, 92.4, 92.8, 96.1, 98.6; IRν: 3 379, 2 946, 2 924, 1 092, 1 026, 1 014 cm-1。
3d:1H NMRδ: 5.44(s, 1H, CH), 5.09(d,J=3.6 Hz, 1H, CH), 4.66(d,J=8.8 Hz, 1H, CH), 4.52(d,J=5.6 Hz, 1H, CH), 3.32~3.63(m, 12H, CH, CH2,糖环-H), 1.27(s, 3H, CH3), 0.90(d,J=6.6 Hz, 3H, CH3), 0.83(d,J=6.3 Hz, 3H, CH3);13C NMRδ: 8.8, 14.5, 18.6, 21.8, 22.7, 23.6, 24.0, 25.0, 27.2, 28.2, 36.8, 62.5, 62.8, 65.0, 67.1, 67.5, 68.1, 69.0, 71.1, 72.1, 72.4, 76.5, 84.3, 92.4, 92.8, 96.1, 98.6; IRν: 3 379, 2 946, 2 924, 1 134, 1 027, 1 061 cm-1。
3e:1H NMRδ: 5.44(s, 1H, CH), 4.98(d,J=6.7 Hz, 1H, CH), 4.91(d,J=3.4 Hz, 1H, CH), 3.49~3.78(m, 6H, CH, CH2,糖环-H), 1.28(s, 3H, CH3), 0.90(d,J=6.5 Hz, 3H, CH3), 0.83(d,J=6.2 Hz, 3H, CH3);13C NMRδ: 8.5, 14.5, 16.6, 18.7, 19.5, 24.8, 27.2, 30.1, 30.8, 30.5, 31.7, 62.5, 68.1, 69.0, 73.1, 72.1, 84.3, 92.4, 92.8, 96.1, 97.9; IRν: 3 380, 2 946, 2 935, 1 135, 1 027, 1 014 cm-1。
2 结果与讨论
2.1 表征
以3a的1H NMR分析为例,δ为5.43, 4.76, 1.28, 0.92和0.83的吸收峰为Ⅱ-H的特征吸收峰;4.90为与Ⅱ-H连接的1a分子端基C1上的H,其J值为6.2 Hz,提示1a端基碳为β-构型。在13C NMR谱中,除溶剂峰外,共有21个信号。青蒿素葡萄糖苷(3a)共有21个C原子,其中Ⅰ母体15个C原子,1a有6个C原子,可确定3a为青蒿素葡萄糖苷。
3c在5.01处的吸收峰为与Ⅱ-H连接的1c分子端基C1上的H,由于其J值非常小,无法以此判断苷键构型,但与3a的糖端基C1上的H相比,化学位移明显向低场移动,推测受邻位羟基的影响,因此1c端基碳亦为β-构型。同理分析1d和1e均为β-构型。
表征结果表明,以α-D-溴代乙酰吡喃糖为糖基给体,采用碱和相转移催化法将Ⅱ引入到糖的C1位差向异构中心合成糖苷时,由于保护基乙酰基的邻位效应使糖的异头碳发生Walden翻转。糖基化反应主要按SN2亲核取代历程进行,因此生成的糖苷中异头碳为β-构型。
2.2 合成及可能的反应机理
由于糖类含有多个羟基,反应活性位点多,通过化学合成方法利用糖定位修饰青蒿素,必须从糖基的保护开始,这也是本合成过程的关键和难点所在。
首先要选择合适的糖基保护方法。最为有效的糖基保护方法主要有乙酰基保护法和异丙叉缩酮保护法。我们比较这两种方法对1a的保护效应,结果发现1a与丙酮在浓硫酸催化剂作用下可得到异丙叉缩酮,但反应条件苛刻,要求在无水环境下进行,目标产物需经柱色谱纯化,操作不便,且目标产物为糖浆状油状物,造成后续反应操作不便。因此本文采用乙酰基保护法,并通过IR谱初步检测发现,保护基团可基本水解掉,不需要额外的脱保护步骤,操作简便。
在糖基保护之后,将糖引入到Ⅱ-H分子中是合成的另一关键步骤。2与Ⅱ-H反应是在碱催化下的水-有机两相反应,其中最为重要是相转移催化剂的选择。本文以2a为例,考察了不同的相转移催化剂[十二烷基三甲基氯化铵(A),十二烷基三甲基溴化铵(B),十二烷基苄基二甲基氯化铵(C),四丁基氯化铵(D),四丁基硫酸氢铵(E)和三乙基苄基氯化铵(F)]对合成3a的影响。结果发现,不同的相转移催化剂对合成3a有较大差异,其中以A, B和C为相转移催化剂时,无3a生成;而以D, E和F为相转移催化剂时,有3a生成,其中以E,即四丁基硫酸氢铵为相转移催化剂时,3a生成量最多,为最理想的相转移催化剂。
[1] Lai C S, Nair N K, Mansor S M,etal. An analytical method with a single extraction procedure and two separate high performance liquid chromatographic systems for the determination of artesunate,dihydroartemisinin and mefloquine in human plasma for application in clinical pharmacological studies of the drug combination[J].Journal of Chromatography B,2007,857(2):308-314.
[2] Zhang D. Determination of artemisinin,artemisinin B and artemisinic acid in herba srtemisiae snnuae by HPLC-UV-ELSD[J].药学学报,2006,42(9):978-981.
[3] Wu H, Moeller K D. Anodic coupling reactions:A sequential cyclization route to the artemisinin ring skeleton[J].Org Lett,2007,9(22):4599-4602.
[4] Efferth T, Volm M. Glutathione-related enzymes contribute to resistance of tumor cells and low toxicity in normal organs to artesunate[J].In Vivo,2005,19(1):225-232.
[5] Liang J, L i Y. Synthesis of the arylether derivatives of artemisinin[J].Chin J Med Chem,1996,6:22-25.
[6] 李玉文,李英霞,张伟,等. 一锅法制备全乙酰吡喃溴代糖[J].有机化学,2004,20(4):438-439.
