Nd掺杂对δ-MoN电子结构和输运性质的影响
2012-09-03尉靖王可答金凤友刘利军张桂玲曾涛
尉靖,王可答,金凤友,刘利军,张桂玲,曾涛
(1.哈尔滨理工大学化学与环境工程学院,黑龙江哈尔滨150080;2.绥化学院食品与制药工程系,黑龙江绥化150026)
钼的氮化物是由氮原子进入金属钼的晶格而形成的一种填隙化合物,这种独特的构造赋予了它优良的物理、化学性质,比如很高的催化活性、耐腐蚀能力、高硬度及超导性等[1-4].钼的氮化物主要有3种存在形式:γ-Mo2N(立方)、β-Mo2N(四方)和δ-MoN(六方),其中γ-Mo2N因其良好的电容特性和类似于贵金属的催化活性而备受人们的关注[5-6].有关δ-MoN的研究主要集中在其高硬度和超导性能上.McMillan曾指出δ-MoN具有与金刚石相比的硬度,Soignard等也曾报道δ-MoN的体弹性模量[7]可达345 GPa[8];δ-MoN 在钼的氮化物中具有最高的超导临界转变温度(Tc),它的Tc因制备条件的不同可以达到12~15 K[9-10].虽然δ-MoN具有很多优异的性能,但有关δ-MoN的报道很少,这主要是由于制备δ-MoN通常需要在高温高压的极端条件下进行,并且在反应产物中常伴有γ-Mo2N、β-Mo2N等杂相,并混有未反应的金属Mo,这就限制了δ-MoN的研究与应用.
最近,李昕等以稀土Nd离子为渗剂,采用化学热扩渗的方法制备了δ-MoN,并且发现Nd离子渗入了δ-MoN体相,大大增强了 δ-MoN的导电性能[11].Nd掺杂导致δ-MoN导电性能的增强说明Nd离子的渗入可能使δ-MoN的电子结构发生了较大变化,特别是Nd离子独特的4f电子构型可能对其产生了较大影响.因此,从理论角度计算Nd掺杂δ-MoN的电子结构,并将其与输运性质的计算结果相结合讨论Nd掺杂对δ-MoN导电性能的影响,探讨其掺杂机理是很有必要的,但这方面理论研究工作尚未见文献报道.
本文从第一性原理出发,利用密度泛函理论(DFT)并结合非平衡格林函数方法,研究了Nd掺杂δ-MoN的电子结构及输运性质,讨论了Nd取代不同位置的Mo原子时对δ-MoN导电性能的影响.
1 模型与计算方法
1.1 VASp计算
δ-MoN具有简单六方结构,其空间群为P63mc(No.186).实验[10]上给出的晶胞参数为 A=5.731 Å,C=5.609 Å.在 Wyckoff坐标系中,Mo原子占据2a(0,0,0)和 6c(0.508,0.492,-0.006)位置,N 原子占据 2b(0.333,0.667,0.299)和 6c(0.164,0.836,0.781)位置,8 个 Mo 原子和 8 个 N原子构成一个δ-MoN原胞,其晶体结构如图1.在图1中标明了各原子所处位置:Mo1和Mo2占据Mo(2a)位置,Mo3~Mo8占据Mo(6c)位置;N1和N2占据N(2b)位置,N3~N8占据N(6c)位置.本文选取Mo2代表Mo(2a)、Mo6代表Mo(6c),分别考虑了Nd取代这2种不同位置的Mo原子时对δ-MoN的晶体结构和电子结构的影响.文中这2类掺杂体系分别称为Nd-MoN(2a)和Nd-MoN(6c).
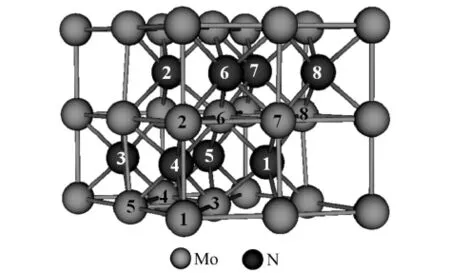
图1 δ-MoN的晶体结构Fig.1 Crystal structure ofδ-MoN
计算采用基于DFT和投影缀加平面波(PAW)方法的VASP[12]软件包完成,交换关联函数采用广义梯度(GGA)近似下的PW91梯度修正函数.为描述Nd-4f电子之间的库仑作用,采用Liechtenstein等提出的旋转不变方法 (rotationally invariant approach)[13]在GGA中增加了 Hubbard参数 U(GGA+U),这里 U=6.5 eV,J=0.7 eV[14],其中 U 描述4f电子间的局域库仑相互作用,J描述4f电子间的局域交换相互作用.在结构优化和电子结构的计算中,平面波截断能选取为520 eV,k点取值采用Monkhorst-Pack方法的7×7×7网络点,收敛判据设为每个原子受力小于0.01 eV/Å,能量变化小于1×10-5eV,所有计算考虑自旋极化.
1.2 ATK 计算
输运性质计算采用基于非平衡格林函数和DFT的Atomistix ToolKit(ATK)[15]程序包中的两极体系方法完成.图2为计算采用的2类Nd掺杂体系的双探针模型:Ag(111)/Nd-MoN(2a)(001)/Ag(111)和Ag(111)/Nd-MoN(6c)(001)/Ag(111).在实际理论模拟中,这样的模型可分为3个部分,即左、右电极和中心散射区,中心散射区包括4个单位原胞的Nd-MoN(2a)或Nd-MoN(6c),以及左、右电极各两层Ag原子以屏蔽中间原子尺度导体对电极势的影响.电子交换关联势采用 Perdew-Zunger形式的局域密度近似(LDA),所有原子采用单ζ极化基组 (SZP)描述.布里渊区取样用Monkhorst-Pack方案进行,选择k网格点为 3×3×50,能量截断半径取为100 Ry以达到计算效率和精度的平衡.在输运性质计算之前,对双探针体系进行了几何结构优化,原子力收敛标准设为0.06 eV/Å.
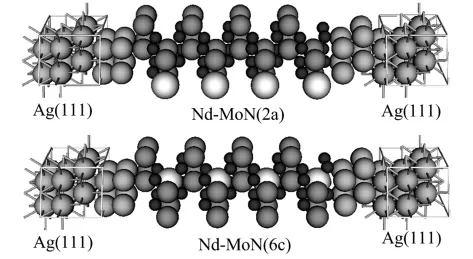
图2 双探针模型示意Fig.2 Schematic of the two-probe systems
2 结果与讨论
2.1 晶体结构
首先对纯δ-MoN的晶体结构进行优化,优化得到的晶胞参数为A=5.777Å,C=5.673Å.定义误差为 (dcalc-dexpt)/dexpt,其中 dcalc和 dexpt分别为计算和实验所得的晶胞参数,A值误差为0.80%,C值误差为1.14%,可以说明本文计算方法的可靠性.对于2类Nd掺杂体系,优化后的晶胞参数列于表1中.
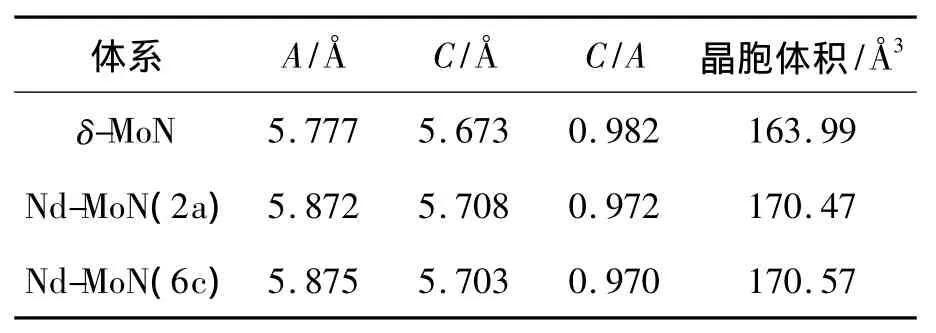
表1 未掺杂及掺杂体系的晶胞参数Table 1 Optimized lattice parameters of the undoped and Nd-dopedδ-moN
从表1中可以看出,Nd掺杂使δ-MoN的晶胞发生膨胀,晶胞参数增大,其中A值变化较大,C值变化较小,导致掺杂后C/A值减小.晶胞膨胀主要是由于Nd的离子半径大于Mo的离子半径造成的(Mo3+的半径为0.68Å,Nd3+的半径为 0.98Å).其中Nd-MoN(2a)和Nd-MoN(6c)的晶胞参数未发现明显差别.
为了进一步说明Nd掺杂对δ-MoN晶胞参数的影响,表2给出了掺杂前被取代 Mo原子及掺杂后Nd原子与其邻近N原子之间的键长.通过比较可以发现,掺杂后的Nd—N键长明显大于掺杂前的Mo—N键长.比如,当Nd取代Mo2时,Nd2—N键的平均键长为2.32Å,而掺杂前Mo2—N键的平均键长只有2.20Å.因此,当Nd原子取代δ-MoN原胞中的Mo原子时,Nd原子与其邻近原子之间的距离增大,导致局域晶格体积膨胀,从而使晶胞参数增大.掺杂的难易程度可以通过形成能来判断,形成能越低,晶体的稳定性越好,越容易掺杂.掺杂的形成能 Eform可由下式计算[16]:

表2 未掺杂及掺杂体系的键长Table 2 Optimized bond lengths of the undoped and Nd-dopedδ-moN
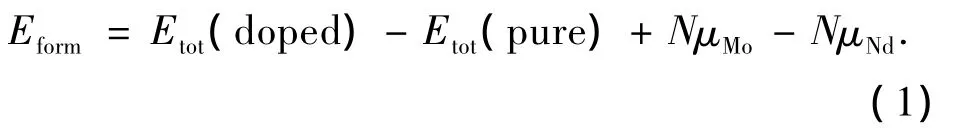
式中:Etot(pure)和Etot(doped)分别为掺杂前后原胞的总能量,μMo和 μNd分别为 Mo和 Nd的化学势,N代表被Nd原子取代的Mo原子数量.由于计算中采用一个Nd原子取代δ-MoN原胞中的一个Mo原子,N=1.对于Nd-MoN(2a)和Nd-MoN(6c),计算得到的形成能分别为1.51 eV、1.54 eV,这表明Nd更易于取代2a位置的Mo原子.
2.2 电子结构
首先采用优化后的晶胞参数计算了未掺杂及掺杂体系的能带结构.在计算中,要画出所有波矢对应的能级是非常困难的,因此,能带结构的计算结果常以图示的形式在第一布里渊区中的一些高对称点、线上给出.δ-MoN属于简单六方结构,其高对称性点的坐标为:Γ(0,0,0)、A(0,0,1/2)、M(1/2,0,0)、K(1/3,1/3,0)、L(1/2,0,1/2)和 H(1/3,1/3,1/2).图3给出了未掺杂及2类Nd掺杂体系沿这些高对称点的能带结构,计算中选择费米能级EF作为能量零点.从图中可以看出,3个体系的价带和导带相互交错重叠,有多条能带与费米能级相交,都呈现出金属性.与纯δ-MoN的能带结构相比,2类Nd掺杂体系的价带与导带数目明显增多,并且变得非常密集,这主要是由于Nd掺杂导致晶胞中的原子位置相对调整,并且Nd掺杂引入了新的能级,电子数目发生改变造成的.
通过对态密度的分析可以更加清楚的看到Nd掺杂对δ-MoN电子结构的影响.图4给出了未掺杂及2类Nd掺杂体系的总态密度(TDOS)和分波态密度(PDOS).从图中可看出,纯δ-MoN的态密度主要由Mo-4d和N-2p轨道电子组成.在低能区(-8.0~ -3.5 eV),态密度由Mo-4d和N-2p轨道电子共同贡献,并且二者的主要峰值相对应,这说明在Mo-4d和N-2p之间存在强烈的杂化效应;而在费米能级附近及高能区 (-3.5~6.0 eV),态密度则主要源于Mo-4d轨道电子的贡献,这说明在δ-MoN中存在着较强的金属键,强的金属键是其具有优良导电性的主要原因.当Nd原子取代δ-MoN中的Mo原子后,Mo-4d和N-2p轨道电子对态密度的贡献未发生明显变化,与未掺杂的情况相比只是在细节上有所差别,态密度的变化主要来自于Nd-4f轨道电子的贡献.由于稀土元素的4f轨道电子具有强关联性,Nd-4f轨道电子的态密度峰发生分裂,分别位于-4~-1 eV、2~4 eV及费米能级附近,其中位于2~4 eV的态密度峰最为显著,但由于其局域性较强且距离费米能级较远,因此对体系的导电性影响不大.这里值得注意的是Nd-4f轨道电子在费米能级附近贡献的态密度峰,它使费米能级附近的态密度增大,导致费米面电子出现的几率增加,由此推测Nd掺杂可以增强δ-MoN的导电性.通过比较可以发现,Nd-MoN(6c)在费米能级附近的态密度要大于Nd-MoN(2a),这说明当Nd取代6c位置的Mo原子时更有利于增强δ-MoN的导电性,这一点可以通过下面的输运性质计算得到证实.

图3 未掺杂及掺杂体系的能带结构Fig.3 Band structures of the undoped and Nd-doped δ-moN
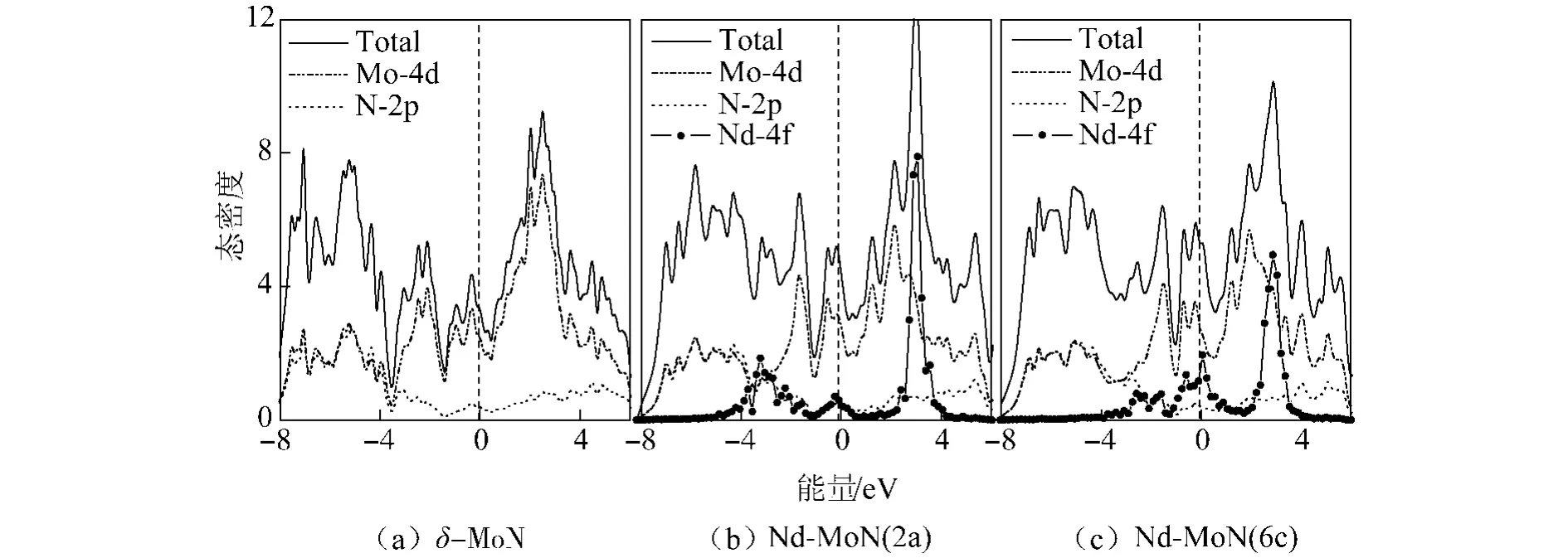
图4 未掺杂及掺杂体系的态密度Fig.4 Density of states of the undoped and Nd-doped δ-moN
2.3 输运性质
体系的输运性质计算采用单电子散射理论[17]来进行,当两电极施加外部偏压V时,通过体系的电流I为
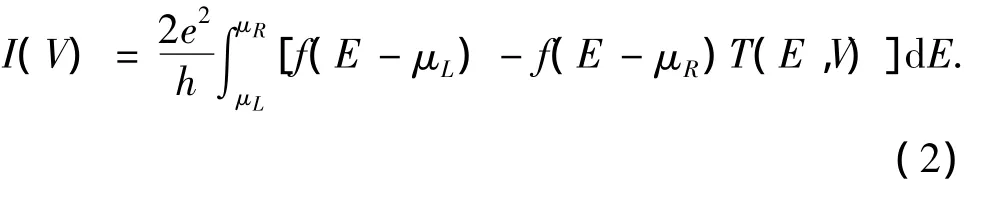
式中:μL,R=EF∓eV/2 为左、右电极的化学势,f为费米分布函数,T(E,V)为电子从一个电极流向另一个电极的穿透函数,其计算公式为
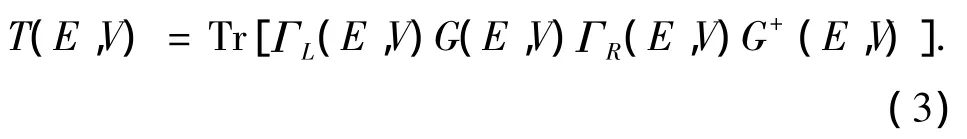
式中:Tr表示矩阵求迹,G和G+为分子修正过的格林函数,ΓL和ΓR为左、右电极与分子之间的耦合作用,它可通过下式计算:
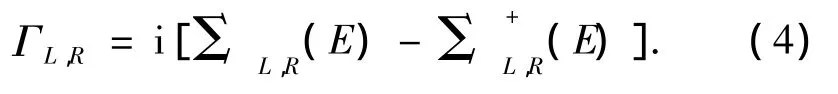
式中:ΣL,R(E)和Σ+L,R(E)为分子对散射区域的自能.通过上述公式,即可计算体系的伏安特性曲线.
图5为未掺杂及2类Nd掺杂体系在偏压范围-1.0~1.0 V的伏安特性曲线.从图中可以看出,3个体系的电流随偏压都呈线性增加,表现出近似的欧姆关系.在相同偏压下,电流由大到小的顺序为Nd-MoN(6c)、Nd-MoN(2a)、δ-MoN,说明 Nd 掺杂可以增强δ-MoN的导电性.其中当Nd原子取代6c位置的Mo原子时,δ-MoN的导电性增强较大,这与前文电子结构分析的结果相符合.
为了进一步说明Nd掺杂对δ-MoN输运性质的影响,计算了掺杂前后的透射谱.根据式(2),体系的电流与透射函数T(E,V)密切相关,只有能量在[μL,μR]区间的电子才对电流有贡献,这个能量区间称为偏压窗口或积分窗口.考虑到费米能级EF已设为 0 eV,偏压窗口[18]实际上就是[-V/2,V/2].因此,体系的电流由偏压窗口内T(E,V)的积分面积所决定,积分面积越大,电流就越大,反之亦然.图6为未掺杂及2类 Nd掺杂体系在0、±1.0 V偏压下的透射谱,在图中标明了偏压窗内的积分面积S.从图中可以看出,Nd掺杂使透射峰的形状和位置发生变化.当偏压为零时,2类Nd掺杂体系在费米能级附近的透射峰的强度要明显大于未掺杂体系,其中Nd-MoN(6c)的透射峰的峰值较高,说明电子遂穿的机率较大.当施加外偏压后,Nd掺杂使偏压窗内的积分面积增大,偏压窗内的S值由大到小顺序为 Nd-MoN(6c)、Nd-MoN(2a)、δ-MoN,这与图5中电流大小的顺序相一致.比如,当偏压为1.0 V 时,δ-MoN、Nd-MoN(2a)和 Nd-MoN(6c)在偏压窗内的积分面积分别为0.338、0.550 和0.567,对应的电流分别为30.80、40.94 和44.41 μA.
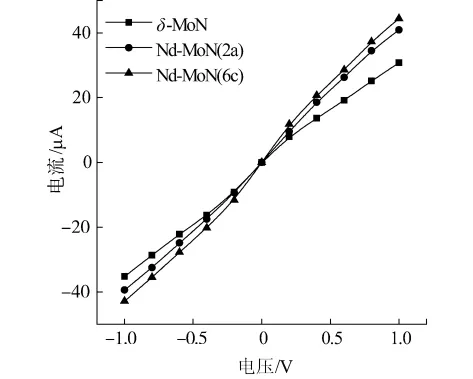
图5 未掺杂及掺杂体系的伏安特性曲线Fig.5 Current-voltage curves of the undoped and Nddopedδ-moN
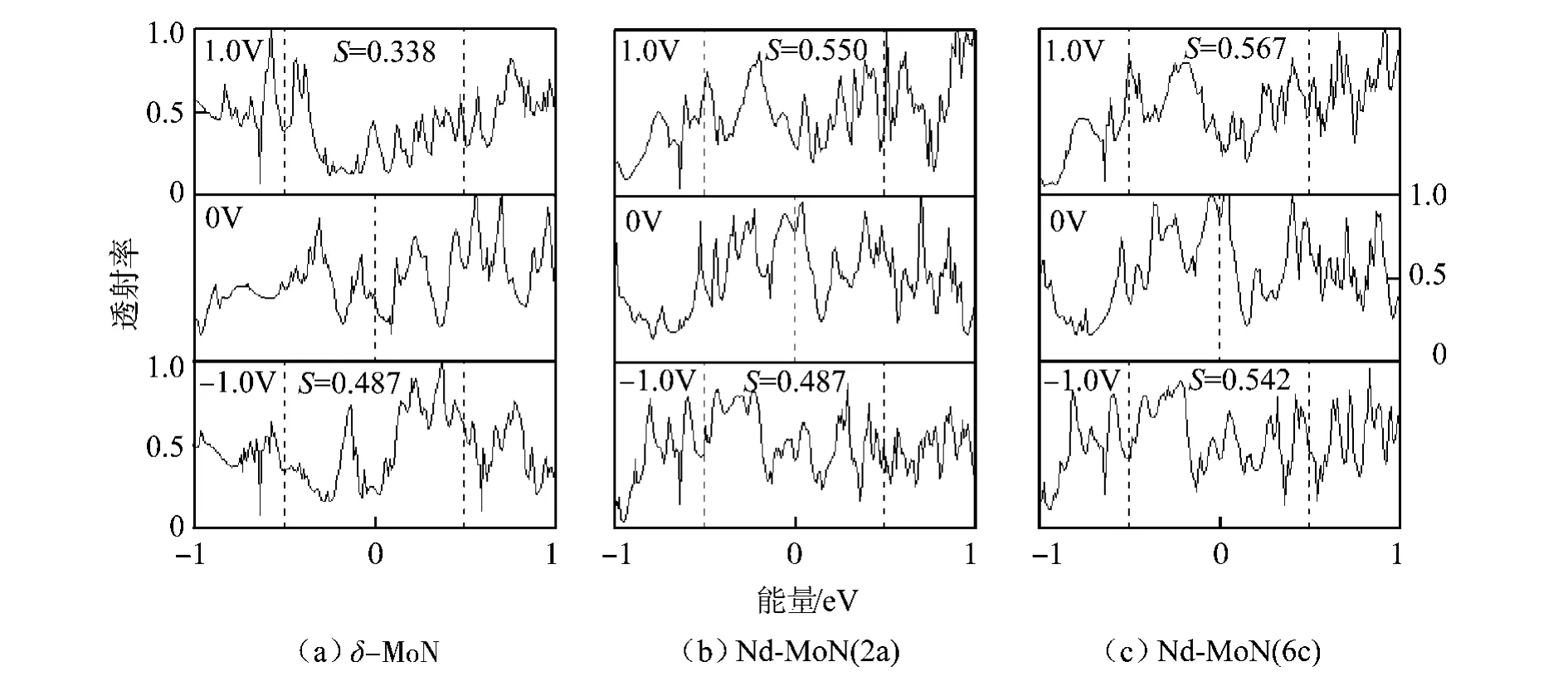
图6 未掺杂及掺杂体系的透射谱Fig.6 Transmission spectra of the undoped and Nd-doped δ-moN
3 结束语
运用密度泛函理论并结合非平衡格林函数方法,计算了Nd掺杂δ-MoN的电子结构和输运性质,讨论了Nd取代不同位置的Mo原子时对δ-MoN导电性能的影响.结果表明,Nd掺杂使费米能级附近的态密度增大,导致费米面电子出现的几率增加,从而增强了δ-MoN导电性,其中当Nd取代6c位置的Mo原子时对δ-MoN的导电性增强较大.该研究有助于了解掺杂对材料导电性影响的机理,并指导设计有实用性能的新型导电材料.
[1]KOJIMA R,AIKA K.Molydenumnitride and carbide catalysts ammonia synthesis[J].Applied Catalysis A:General,2001,219(1):141-147.
[2]LI X,TANG B,PAN J,et al.Tribological properties of Mo-N hard coatings on Ti6Al4V by double glowdischarge technique[J].Journal of Materials Science & Technology,2003,19(4):291-293.
[3]ANITHAA V P,VITTAB S,MAJORA S.Structure and properties of reactivity sputtered γ-Mo2N hard coatings[J].Thin Solid Films,1994,245(1/2):1-3.
[4]GOMATHIA,SUNDARESAN A,RAO C N R.Nanoparticles of superconducting γ-Mo2N and δ-MoN[J].Journal of Solid State Chemistry,2007,180(1):291-295.
[5]朱云贵,李学良,何建波,等.铝对氮化钼膜电极性能的影响[J].应用化学,2001,18(7):54-60.ZHU Yungui,LIXueliang,HE Jianbo,et al.The effect of alumimiumon the properties ofmolybdenumnitride filmelectrode[J].Chinese Journal Applied Chemistry,2001,18(7):54-60.
[6]NEYLON mK,BEJSK,BENNETT C A,et al.Ethanol amination catalysis over early transition metal nitrides[J].Applied Catalysis A:General,2002,232(2):13-17.
[7]MCMILLAN pF.Newmaterials fromhigh-pressure experiments[J].Nature Materials,2002,1:19-25.
[8]SOIGARD E,MCMILLAN pF,CHAPLIN T D,et al.High-pressure systhesis and study of low-compressibility molybdenumnitride(MoN and MoN1-x)phases[J].Physical ReviewB,2003,68(13):132101.
[9]BEZINGE A,YVON K,MULLERW,etal.High-pressure high-temperature experiments on δ-MoN[J].Solid State Communication,1987,63(2):141-145.
[10]BULL C L,MCMILLAN pF,SOIGNARD E.Structure determination of delta-MoN prepared at high pressure and high temperature by neutron and X-ray diffraction[J].Journal Solid State Chemistry, 2004, 177(4/5):1488-1492.
[11]LIX,LIZ H,GAO Y Z,et al.Conductivity of(NH4)3[CrMo6O24H6]·7H2O treated by Nd chemistry-heated diffused permeation[J].Journal of Materials Science &Technology,21(5):776-778
[12]MADSEN G K H,BLAHA P,SCHWARZ K,et al.Efficient linearization of the augmented plane-wave method[J].Physical ReviewB,2001,64(19):195134.
[13]LIECHTENSTEIN A I,ANISIMOV V I,ZAANEN J.Density-functional theory and strong interactions:Orbial ordering in Mott-Hubbard insulators[J].Physical ReviewB,1995,52(8):R5467-R5470.
[14]ANISIMOV V I,ARGASETIAWAN F,LICHTENSTEIN A I.First-principles calculations of the electronic structure and spectra of strongly correlated systems:the LDA+U method[J].Journal Physics:Condensed Matter,1997,9(4):767-808.
[15]QuantumWise.Atomistix ToolKit version 2.0,Quantum-Wise A/S[EB/OL].[2012-3-2].www.quantumwise.com.
[16]CANTELE G,DEGOLIE,LUPPIE,et al.First-principles study of n-and p-doped silicon nanoclusters[J].Physical ReviewB,2005,72(11):113303.
[17]DATTA S.Electronic transport in mesoscopic systems[M].Cambridge:Cambridge University Press,1995:1-377.
[18]BRANDBYGEM,MOZOSJL,ORDEJÓN P,etal.Density-functionalmethod for nonequilibriumelectron transport[J].Physical ReviewB,2002,65(16):165401.
