一种莱茵衣藻启动子功能检测系统的构建
2012-07-16王潮岗黄惠珠孙海珊胡章立雷安平
王潮岗,黄惠珠,孙海珊,胡章立,雷安平
深圳大学生命科学学院,深圳市海洋生物资源与生态环境重点实验室,深圳518060
莱茵衣藻(Chlamydomonas reinhardtii)是单细胞、带鞭毛的真核光合生物,因具有培养简单、生长周期短、光合效率高和遗传转化技术成熟等优点,广泛用于生物学基础研究[1-2].它可以在细胞中表达各种异源蛋白,如荧光素酶[3]和 SEC2蛋白[4]等;也可用于启动子功能的研究,如在莱茵衣藻中进行了 Rbcs2 启动子[5]和 Hsp 70A 启动子[6]功能的研究.但微藻启动子功能研究进展依然缓慢.究其原因,与未建立便捷、有效的研究系统直接相关.
目前,研究微藻启动子功能的手段包括缺失突变和点突变等,它们都需要合适的T载体辅助.缺乏商业化的微藻克隆、表达T载体,阻碍了微藻功能基因和相关启动子功能的研究.T载体一般包括克隆T载体和表达型T载体,一般通过平末端加T法和内切酶法获得[7-8].T载体末端带有突出的T碱基,可直接与Taq酶扩增产物末端突出的A碱基配对,提高了聚合酶链式反应 (polymerase chain reaction,PCR)产物与载体的连接效率,促进对基因克隆与表达的研究[9].然而,国内藻类启动子的研究主要集中在与光合作用、固氮作用相关基因的启动子上[10],如 Rubisco 启动子[11-12]和 cpc 启动子[13-14]等,可检测藻类启动子功能的T载体鲜有报道.笔者认为,通过PCR获得启动子5'或3'端缺失片段,连接到T载体,转化到成熟的表达系统,再通过筛选基因或报告基因分析启动子功能,可实现对其快速检测.
莱茵衣藻作为研究真核微藻的模式生物,因其清晰的遗传背景和成熟的遗传转化体系,具有开发成微藻启动子功能检测系统的潜力.本研究尝试构建莱茵衣藻启动子功能检测T载体,并将PCR获得的雨生红球藻β-胡萝卜素酮化酶基因bkt1启动子[15]片段直接克隆到T载体上,在莱茵衣藻中验证启动子的功能,构建了一种新的检测微藻启动子功能的方法,为研究微藻启动子开辟了新的途径.
1 材料与方法
1.1 质粒、菌株与衣藻
质粒091127-a3由本实验室筛选雨生红球藻基因组文库所得,测序分析发现其含有bkt1上游调控序列.质粒pSP124由英国伦敦大学Saul Purton教授惠赠,携带ble基因 (使宿主细胞获得腐草霉素和Zeomycin抗性)、Rbcs2启动子和终止子.载体pMD18-T购自Takara Biotechnology(大连)有限公司.
大肠杆菌(Escherichia coli,E.coli)菌株XL1由本实验室保存.
莱茵衣藻CC-849由美国杜克大学Chlamydomonas Genetic Center提供,培养在Tris-acetate-phosphate(TAP)培养基中.
1.2 试剂和仪器
质粒提取试剂盒、DNA片段纯化试剂盒和LA Taq酶 (含 dNTP、Taq酶和缓冲液)等均购自Takara Biotechnology(大连)有限公司;丙烯酰胺和TEMED等电泳试剂均购于Bio-Rad公司;十二烷基硫酸钠 (sodium dodecyl sulfate,SDS)、Tris碱和EDTA等购自生工生物工程 (上海)股份有限公司.以上试剂均为分析纯.
实验所用PCR仪为MJ-Research公司的PTC-100;冷冻高速离心机为Eppendorf公司的Centrifuge 5804 R;杂交箱为HL-2000 HybriLinker.
1.3 引物设计
根据091127-a3质粒上bkt1的上游调控序列设计引物pB-bkt4和pB-bkt5;根据pSP124质粒上的ble设计引物pBle1和pBle2;根据bkt1上游调控序列设计引物pcrto1、pcrto2、pB-bkt7和pB-bkt8,引物pB-bkt7和pB-bkt8引入了Eam 1105Ⅰ、EcoRⅤ和BamHⅠ位点;根据ble序列设计引物PrBle1和PrBle2,具体见表1.
1.4 莱茵衣藻启动子功能检测T载体的构建
pB124载体的构建.以pBle1和pBle2为引物,通过PCR从质粒pSP124中扩增出大小为970碱基对 (base pair,bp)的ble-3'-Rbcs2终止子 (扩增条件为94℃ 变性3 min;94℃反应1 min,58℃反应1 min,72℃反应1 min,25个循环;72℃ 延伸10 min),克隆到pMD 18-T载体中,获得T-p124质粒.通过EcoRⅤ和KpnⅠ双酶切质粒T-p124获得ble-3'-Rbcs2终止子,将其插入pSP124载体的EcoRⅤ和KpnⅠ位点,获得pB124载体.

表1 引物列表Table 1 List of primer sets
pB载体的构建.通过Eam1105Ⅰ酶切pB124载体,获得线性载体,再经T4 DNA Polymerase使载体末端平滑化,突变原有的Eam1105Ⅰ位点,通过T4连接酶连接载体,获得pB载体.
pB-0.45T载体的构建.以pB-bkt4和pB-bkt5为引物,从091127-a3中扩增出3 735 bp的bkt1上游调控序列,克隆到pMD 18-T载体获得T-5'bkt.以pB-bkt9和pB-bkt10为引物,通过PCR从质粒T-5'bkt中扩增出450 bp的启动子序列,获得引入Eam1105Ⅰ、EcoRⅤ和BamHⅠ位点的启动子序列,命名为5'bkt1-0.45.通过EcoRⅤ和BamHⅠ双酶切5'bkt1-0.45,将其插入pB载体的EcoRⅤ和BamHⅠ位点,最后获得pB-0.45T载体,见图1.
1.5 启动子序列的T克隆
T载体的制备.采用 Eam1105Ⅰ 双酶切 pB-0.45T载体,于37℃反应1 h,酶切产物经琼脂糖凝胶 (琼脂糖质量浓度为10 g/L)电泳分离,回收的大片段即为T载体,经DNA片段纯化试剂盒纯化后可直接应用于PCR片段的T克隆.
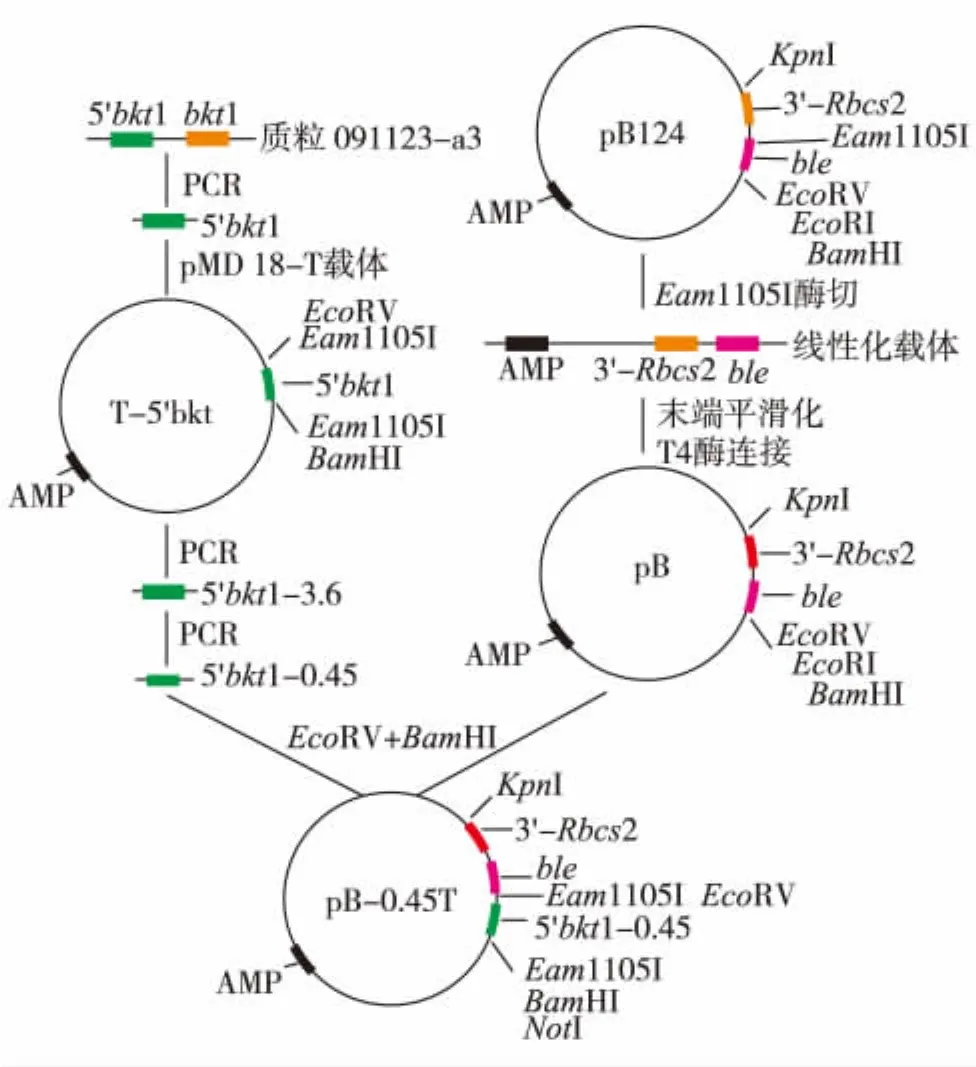
图1 pB-0.45T检测T载体的构建Fig.1 Construction of pB-0.45T
启动子片段的T克隆.利用引物pcrto1和pcrto2,从质粒091127-a3中扩增出长度为1 986 bp的bkt1启动子片段,与T载体混匀,T4连接酶连接后转化到感受态大肠杆菌XL1细胞中,经氨苄抗生素筛选可获得转化子,经T3和T7引物测序验证 (深圳华大基因研究院),命名为pB-2T.
1.6 莱茵衣藻的遗传转化
莱茵衣藻遗传转化采用“珠磨法”[16].诱导时,加入3 mol/L的NaAC至终浓度为45 mmol/L.单个细胞遗传转化效率计算公式为

1.7 衣藻基因组DNA提取
微藻总DNA提取参照文献[16]的方法,如采用Qiagen®公司的Puregene Tissue Core Kit A提取微藻总DNA,则所有操作按产品说明书进行.
1.8 转基因衣藻的DNA-PCR检测
以提取的莱茵衣藻基因组DNA为模板,进行PCR扩增检测.使用特异性引物PrBle1和PrBle2扩增ble基因片段 (464 bp),Act1和Act2扩增βactin基因片段 (852 bp).PCR反应条件为:94℃变性5 min;94℃反应1 min,58℃反应90 s,72℃反应90 s,30个循环;72℃延伸10 min.
1.9 转基因衣藻的Western杂交
将培养至对数生长期后期的藻细胞进行处理,取藻液25 mL,于4℃,5 000 r/min离心3 min,弃上清;加入1 mL PBS重悬藻细胞,小心加入0.5 mL玻璃珠 (Biospec 11079105),加盖,冰上放置5 min;使用机械破碎机 (Mini-Beadbeater,Biospec)进行破碎,每次以最大速度破碎25 s,冰浴1 min,重复5次;然后,于4℃,12 000 r/min离心20 min,小心收集上清,上清即为藻细胞总蛋白.
微藻总蛋白经聚丙烯酰胺凝胶电泳 (SDS-polyacrylamide gel electrophoresis,SDS-PAGE)分离后,通过半干式电转仪 (Trans-Blot SD,Bio-Rad,美国)转移到硝酸纤维膜 (Hybond C-super,Amersham)上.显色根据产品说明书进行操作 (BCIP/NBT,aMRESCO®099OC099,美国).BLE多克隆抗体购自法国Invivogen公司,内参使用鼠抗β-Tubulin单抗 (EarthOx),作为二抗的羊抗兔IgG和羊抗鼠IgG购自Proteintech公司.
2 结果与分析
2.1 莱茵衣藻启动子功能检测T载体的构建
其构建思路为:首先,利用PCR和酶切技术去除pSP124载体上的Rbcs2启动子;其次,利用T4 DNAPolymerasep突变启动子上面的一个Eam1105Ⅰ位点,以bkt1启动子的450 bp序列为间隔序列,并通过PCR引入了两个Eam1105Ⅰ位点,它的识别序列是5'-GACTTTACGTC-3',从3'第5个碱基开始水解,因此Eam1105Ⅰ双酶切恰好在5'末端获得两个突出的T碱基,可与PCR产物3'末端突出的A碱基互补;最后,经过T4连接酶的连接即可将PCR产物直接连到载体上,形成完整的ble表达框,在莱茵衣藻中表达BLE蛋白 (使宿主细胞获得腐草霉素和Zeomycin抗性),并通过Zeomycin筛选出转基因藻,见图2.

图2 pB-0.45T的T载体示意图Fig.2 Diagram-0.45T
pB-0.45T载体的鉴定.因 pB-0.45T载体由5'bkt1序列插入pB载体的BamHⅠ和EcoRⅤ位点得到,所以,它以450 bp的5'bkt1序列为间隔序列.采用引物pB-bkt9和pB-bkt10,可从pB-0.45T载体中扩增出450 bp的目的条带 (图3),表明pB-0.45T载体含有450 bp的5'bkt1.通过Eam1105Ⅰ双酶切pB-0.45T,得到长度为450 bp和约3 000 bp的片段,通过EcoRⅤ和KpnⅠ双酶切得到970 bp和约 3 000 bp的片段 (图4),表明 450 bp的5'bkt1序列是正向插入pB载体的.以上验证与pB-0.45T载体的构建吻合,表明已成功引入了两个Eam1105Ⅰ限制性酶切位点,由450 bp的bkt1启动子、ble和Rbcs2终止子组成了完整的ble表达框(图2),莱茵衣藻外源启动子功能检测T载体已构建成功.

图3 pB-0.45T的PCR鉴定Fig.3 PCR analysis of pB-0.45T
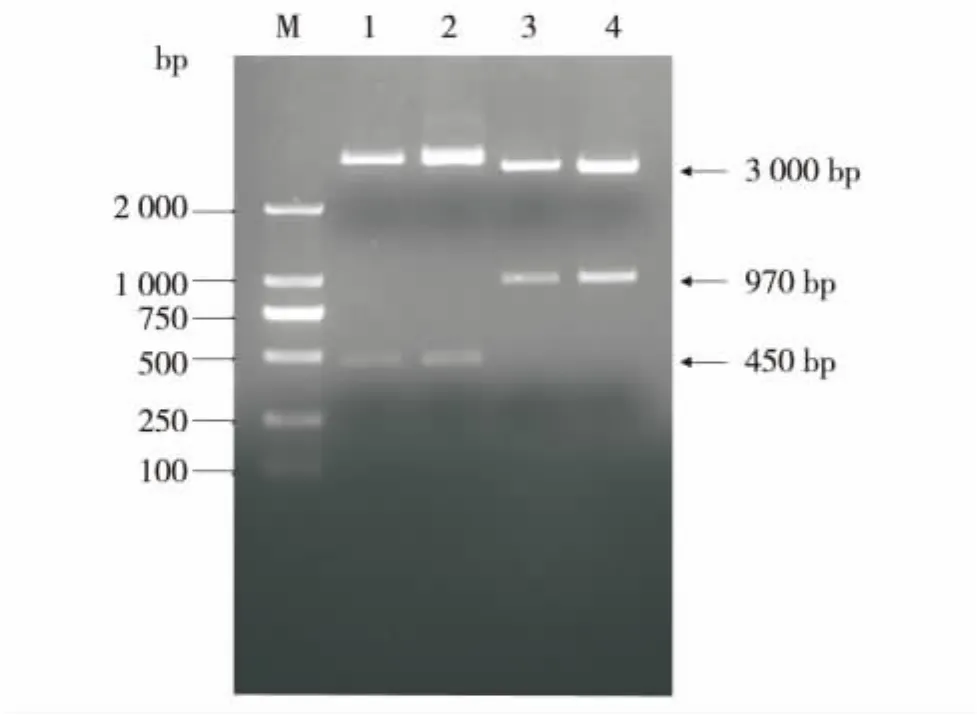
图4 pB-0.45T的酶切鉴定Fig.4 Digesting pB-0.45T with restrictive enzyme
2.2 启动子序列的T克隆
pB-0.45T属于穿梭载体,具有氨苄青霉素抗性,可转化到大肠杆菌中筛选重组子.同时,为方便于启动子活性分析,引入两个Eam1105Ⅰ位点来构建T载体,通过Eam1105Ⅰ酶解获得插入PCR产物的T克隆位点 (图2).本研究通过PCR获得bkt1启动子 (1 986 bp)的5'缺陷序列,插入pB-0.45T载体的T克隆位点.PCR结果显示,1 986 bp的bkt1启动子序列已成功插入pB-0.45T中,pB-2T检测载体已构建成功,见图5.
2.3 转基因藻的获得
通过“珠磨法”将pB-0.45T和pB-2T检测载体转化入细胞壁缺陷的莱茵衣藻CC-849中,获得的转基因衣藻分别命名为TranB-0.45和TranB-2.同时建立一个阳性对照,将质粒pSP124转化进入莱茵衣藻CC-849中,获得的转基因衣藻命名为TranBle.
遗传转化结果显示:TranB-0.45未加醋酸钠时得不到转化子,加入后则单个细胞遗传转化效率提高到1.67×10-8,与 TranBle的转化效率接近,而TranBle上使用的Rbcs2启动子是莱茵衣藻转化效果最好的启动子之一,表明bkt1启动子的450 bp序列也可以用于莱茵衣藻转化.TranB-2未能获得转化子,可能是该启动子片段含有抑制外源基因整合的调控元件.各转基因藻遗传转化结果见表2.

图5 pB-2T载体的PCR鉴定Fig.5 PCR analysis of pB-2T

表2 转基因藻的遗传转化效率Table 2 Transformation frequency of transgenic algae
2.4 转基因藻的DNA-PCR鉴定
提取转基因衣藻TranB-0.45、TranBle和莱茵衣藻CC-849的基因组DNA,利用PrBle1、PrBle2引物对和Act1、Act2引物对检测转基因藻.结果显示,转基因藻TranB-0.45和TranBle均能扩增出464 bp的ble片段和852 bp的β-actin基因片段,与阳性对照结果一致.转基因藻均在TAP平板 (含10 μg/mL Zeomycin)上培养,经多次传代后再提取基因组DNA,PCR仍可扩增出ble片段(图6),证明ble已经整合到莱茵衣藻基因组中,并可稳定传代.
2.5 各插入片段的启动子功能分析
离心收集培养至对数后期的转基因衣藻TranB-0.45和TranBle,提取其总蛋白,BLE抗体杂交,以检测转基因藻中BLE的表达情况.其中,TranBle为阳性对照,莱茵衣藻CC-849为阴性对照.结果显示,转基因衣藻TranB-0.45能正确表达BLE蛋白,在13.6 kD处显示清晰的杂交印迹,与阳性对照一致,表明450 bp的bkt1启动子片段可以正确表达BLE蛋白,具有启动子活性 (图7).以上结果表明,莱茵衣藻及本研究所构建的启动子功能检测T载体所组成的检测系统是有效的.
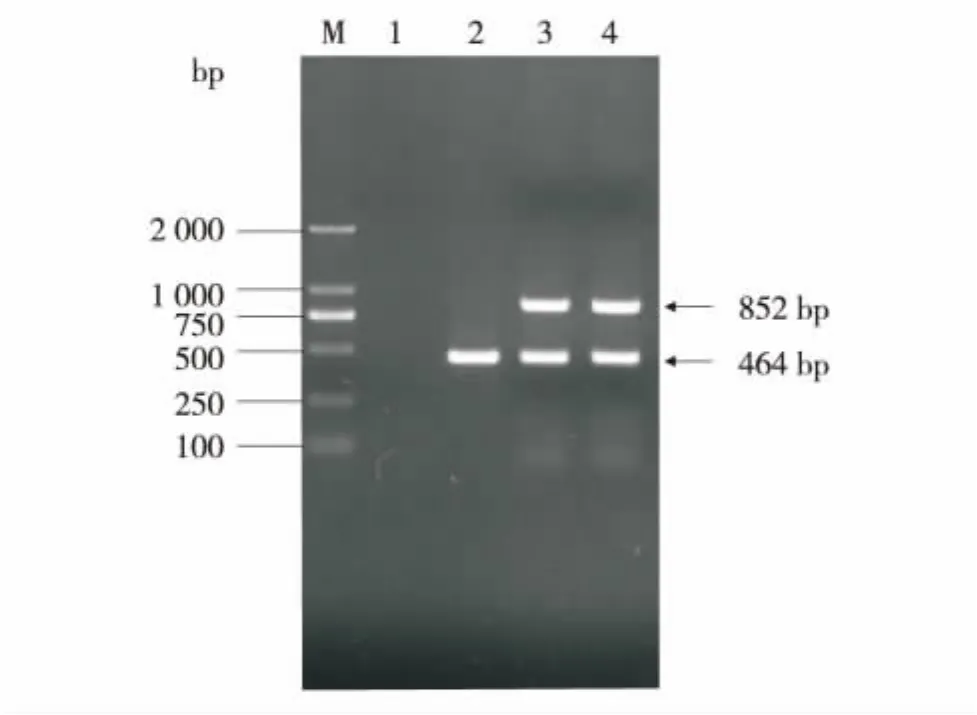
图6 转基因衣藻的PCR检测Fig.6 PCR analysis of transgenic algae

图7 BLE蛋白在转基因衣藻中的表达Fig.7 Expression of BLE in transgenic algae
3 讨论
T载体是一种方便、高效的克隆载体,是基因工程领域重要的研究工具.它可直接将PCR产物连接到载体上,通过测序获得序列信息,也可直接用于目的基因的表达研究[17].目前,已有许多商业化的克隆T载体,如pMD 18-T(Takara),表达型的T载体则还不太成熟,仅有少数公司提供,如pTARGET载体 (Promega)和pGW-T载体 (Qiagem).可用于启动子功能检测的载体已有研究,但未见任何商业性的T载体,很多实验仍通过常规的PCR扩增、克隆和酶切连接到特定的载体上,转化到宿主细胞后再进行检测,该方法工作量大、效率低,用于研究微藻启动子就更困难了.因缺少针对微藻研究的商业化T载体,大大阻碍了微藻基因工程的研究.因此,建立一个拥有表达型T载体的微藻启动子功能研究系统具有重要科学价值.
通过限制性内切酶获得可克隆PCR片段的T克隆位点,该方法具有方便、稳定的优点,是设计T载体的先进手段[18].利用XcmⅠ双酶切获得的T载体具有高效、低背景的优点[17].同样,利用Eam1105Ⅰ双酶切获得T载体的技术可以满足高通量克隆和表达基因的需要[19].本研究采用Eam1105Ⅰ双酶切获得的T载体效率高,根据TA克隆的原理,插入的外源启动子序列与ble、Rbcs2终止子组成完整的ble表达框.但是,T克隆载体也有弱点,太长的片段不易被直接克隆,而启动子由于具有复杂的功能,序列有时会非常长,因此,pB-0.45T载体上还设计了BamHⅠ和EcoRⅤ限制性内切酶位点,可以满足大多数启动子的克隆与功能研究.如插入的启动子片段具有活性,正确表达BLE蛋白,则转基因衣藻细胞因具有Zeomycin抗性而被筛选出来;如不能表达BLE蛋白则不能获得转化子.由于莱茵衣藻对Zeomycin非常敏感,本研究未检测到假阳性.研究表明,450 bp的bkt1启动子具有与莱茵衣藻Rbcs2启动子同样能力,在莱茵衣藻中表达异源蛋白.pB-0.45T载体和莱茵衣藻适合作为微藻启动子功能研究的有效平台,有望推动微藻启动子功能研究.
/References:
[1]Harris E H.The Chlamydomonas Source Book:A Comprehensive Guide to Biology and Laboratory Use[M].SanDiego(USA):Academic Press,1989.
[2]WANG Chao-gang,HU Zhang-li.The research advance in nuclear transformation system of Chlamydomonas reinhardtii[J].Journal of Zhongkai University of Agriculture and Technology,2005,18(2):59-64.(in Chinese)王潮岗,胡章立.莱茵衣藻细胞核转化系统研究进展[J].仲恺农业技术学院学报,2005,18(2):59-64.
[3]Ning Shao,Ralph Bock.A codon-optimaized luciferase from Gaussia princeps facilitates the in vivo monitoring of gene expression in the modelalga Chalmymononas reinhardtii[J].Current Genetics,2008,53:381-388.
[4]LI Jian-cheng,PENG Shi-qing,HU Zhang-li.Expression and immunologicalactivity of enterotoxin C2 from Staphylococcusaureus in Chlamydomonasreinhardtii[J].Journal of Shenzhen University Science and Engineering,2012,29(2):159-164.(in Chinese)李建成,彭世清,胡章立.SEC2在莱茵衣藻中表达及免疫学活性分析[J].深圳大学学报理工版,2012,29(2):159-164.
[5]Stevens D R,Rochaix J D,Purton S.The bacterial phleomycin resistance gene ble as a dominant selectable marker in Chlamydomonas[J].Molecular Genetics and Genomics,1996,251(1):23-30.
[6]Lumbreras V,Stevens D,Purton S.Efficient foreign gene expression in Chlamydomonas reinhardtii mediated by an endogenous intron[J].The Plant Journal,1998,14(4):441-448.
[7]HUANG Wen-jun,WANG Ying.Construction of the high efficiency and low background T vector[J].China Biotechnology,2010,30(12):60-65.(in Chinese)黄文俊,王 瑛.一种高效低背景T载体的构建[J].中国生物工程杂志,2010,30(12):60-65.
[8]LIN Chen-shui,WU Sheng-wei,YANG Dan-yan.Development of T vectors[J].Journal of Zhejiang University of Technology,2009,37(6):623-628.(in Chinese)林陈水,武胜伟,杨丹燕.T载体研究进展[J].浙江工业大学学报,2009,37(6):623-628.
[9]QI Xiang-hui,CHEN Hui,SHEN Qi,et al.Simple construction method of a new T-vector and its application[J].Biotechnology Bulletin,2011(3):163-174.(in Chinese)齐向辉,陈 辉,沈 琦,等.一种构建新型T载体的简便方法及应用[J].生物技术通报,2011(3):163-174.
[10]ZHANG Xue-cheng,GUO Nan,SONG Xiao-jin.Research progress on structure and function of algal promoters[J].Periodical of Ocean University of China,2008,38(3):404-412.(in Chinese)张学成,郭 楠,宋晓金.藻类基因启动子结构与功能研究进展[J].中国海洋大学学报,2008,38(3):404-412.
[11]Yuzuru M,Makoto H,Hitoshi K,et al.Reporter gene introduction and transientexpression in protoplastsof Porphyra yezoensis[J].Journal of Applied Phycology,2004,16(1):23-29.
[12]Walker T L,Becker D K,Collet C.Characterisation of the Dunaliella tertiolecta RbcS genes and their promoter activity in Chlamydomonas reinhardtii[J].Plant Cell Reports,2005,23(10/11):727-735.
[13]Lu Y Z,Zhang X C.Analysis on promoter elements of cpc operon from Arthrospina platensis[J].Acta Oceanologica Sinica,2008,27(1):85-91.
[14]Guo N,Zhang X C,Lu Y Z,et al.Analysis on the factors affecting start-up intensity in the upstream sequence of phycocyanin β subunit gene from Arthrospira platensis by site-directed mutagenesis[J].Biotechnology Letters,2007,29(3):459-464.
[15]HUANG Jun-chao,CHEN Feng,Sandmann G.Stress-related differential expression of multiple β-carotene ketolase genes in the unicellular green alga Haematococcus pluvialis[J].Journal of Biotechnology,2006,122(2):176-185.
[16]WANG Chao-gang,HU Zhang-li,HU Wei,et al.Expression and molecular analysis of phbB gene in Chlamydomonas reinhardtii[J].Chinese Science Bulletin,2004,49(16):1713-1717.
[17]WANG Bao-li,LI Xiao-xia,ZHENG Fang,et al.pEGFPT,a novel T-vector for the direct,unidirectional cloning and analysis of PCR-amplified promoters[J].Biotechnol Letter,2007,29(2):309-312.
[18]ZHANG Chao,LIU Gang,YU Shao-wen,et al.A Secretive Pichia pastoris expression vector for direct PCR product cloning[J].China Biotechnology,2007,27(1):52-58.(in Chinese)张 超,刘 刚,余少文,等.可直接克隆PCR产物的毕赤酵母分泌型表达载体[J].中国生物工程杂志,2007,27(1):52-58.
[19]CHEN Song-biao,Pattavipha Songkumarn,LIU Jian-li,et al.A versatile zero background T-vector system for gene cloning and functional genomics[J].Plant Physiology,2009,150:1111-1121.
