氧化钆掺杂氧化铈的分子动力学模拟
2012-06-23陈云俊孙毅刘一志康高英
陈云俊,孙毅,刘一志,康高英
(哈尔滨工业大学航天科学与力学系,黑龙江哈尔滨150001)
目前,燃料电池作为一种清洁能源越来越受到人们的关注[1].氧化钆掺杂的氧化铈(GDC)是目前应用较为广泛的固体氧化物燃料电池(SOFC)的电解质之一,GDC可以在中低温(500~700 K)下工作,且温度较低时其电导率比传统的YSZ(氧化钇稳定的氧化锆)电解质高一个数量级以上[2].由于工作环境及材料尺度的限制,当前很难用传统的实验手段清晰地揭示GDC在各种工作环境下的体系变化.分子动力学(MD)模拟作为一种在原子尺度下研究材料性能演化规律的技术,被广泛用于GDC的性能研究.目前国际上关于MD模拟GDC的相关工作已经取得很多成就[3-5].虽然在MD模拟过程中,积分算法、时间步、初始构型等因素都能够影响模拟结果,但作为描述模型中粒子间相互作用的函数,势函数才是MD模拟成功的核心因素.当前,采用不同势参数对GDC进行系统MD模拟的工作相对较少,此处总结了当前使用较多的几组势参数,同时给出了一套新的势函数,并对GDC模型进行了系统的模拟,详细解释了模拟过程中体系发生的变化及其导电机理.
1 GDC势函数
GDC是掺杂了氧化钆的氧化铈固溶体,化学式为 CexGd1-xO2-0.5x(x 为 Gd3+的摩尔浓度),其分子动力学模拟是建立在Born固体模型的基础上,阳离子之间只有库伦作用.一般采用 Born-Meyer-Buckingham函数来描述GDC间粒子的相互作用,具体表达式为[6]

式中:qi、qj分别为离子 i与 j的电荷量;rij为 i,j间对应的离子间距;ε0为介电常数;A、ρ、C为对应参数,表1列出了目前较常用的几组参数.其中,Ⅰ:Lewis提出的势参数[6];Ⅱ:Gotte提出的势参数[7];Ⅲ:Licia提出的势参数[8].
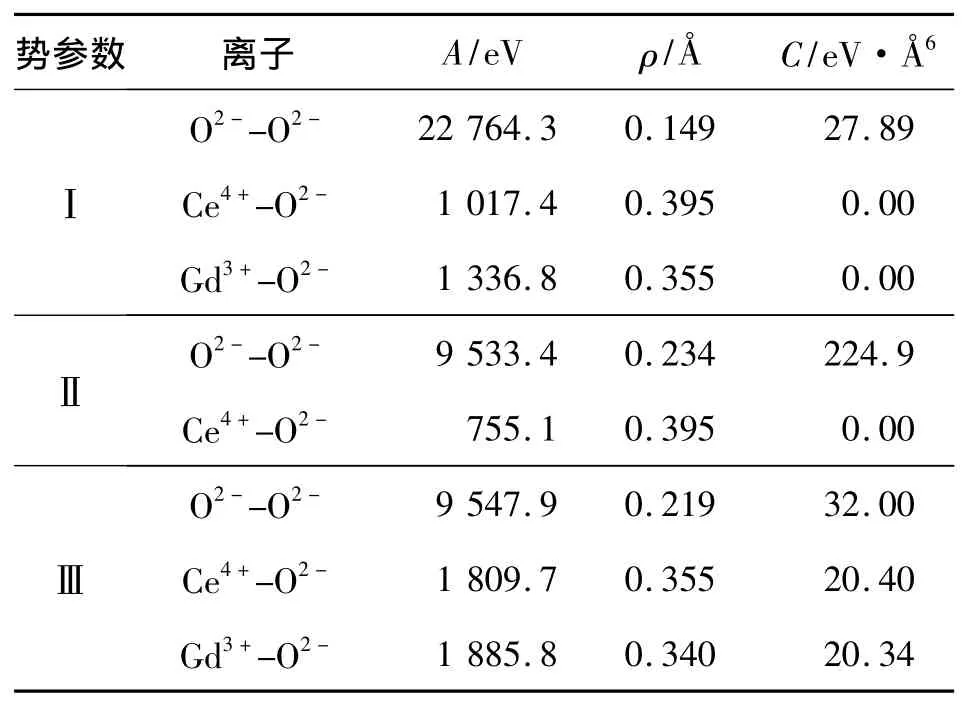
表1 不同势参数列表Table 1 Different potential parameters
表1中的势函数及对应参数主要基于经验势方法,其参数的获得十分依赖实验值,应用范围相对狭窄.在此,依据量子物理和量子化学方法,提出了一种离子晶体势函数的求取方法,应用范围较广.势函数形式与式(1)有所区别,具体如下[9]:

式中:Φ+-描述了阴阳离子之间的作用,Φ++描述了阳离子之间的作用,Φ--描述了阴离子之间的作用.a、b、c为对应的势参数,具体如表2.

表2 新势函数及对应参数Table 2 New potential and corresponding parameters
2 分子动力学模拟
2.1 晶格常数
首先计算了298 K时的晶格常数.模拟过程中,采用NPT系统,时间步为1 fs.先根据晶体学知识建立尺寸为5l×5l×5l(其中l为设定的初始晶格常数,值为5.41Å)的模型,再对整个模型在298 K温度下弛豫100万步,模型完全稳定后,取后50万步的平均值作为计算结果.
图1给出了不同掺杂浓度(文中掺杂浓度都的摩尔浓度)下GDC的晶格常数变化情况.由图1可知,除了Gotte参数的计算结果,其他4组势参数得到的结果与实验值[10]变化趋势一致.实际上只给出了CeO2的势参数,并未给出Gd2O3的参数[7],为了对比不同参数的计算效果,采用Gotte的CeO2参数结合其他文献给出的Gd2O3参数计算了GDC晶格常数.

图1 模拟晶格常数与实验值的对比Fig.1 Calculated lattice constants from different potentials and experimental data
对于没有掺杂的CeO2材料,粒子间通过势函数相互作用,整个体系处于稳定状态.Gd2O3的掺杂引入了氧空位,体系中粒子受力平衡被破坏.为了达到新的平衡状态,粒子的位置发生了改变,整个体系产生膨胀,从而导致晶格常数增加.图1中4组势参数得到的结果对比,可以看出Lewis的结果有较大区别,这是因为该组参数提出于20世纪80年代,没有充足的实验值对参数进行验证,因此该套参数的计算结果与实验值差别较大,只能用于定性说明体系的变化情况;而Hideaki及Licia的势参数是基于大量实验数据基础上拟合得到的,因此这2套参数的计算值较为接近实验值,更能反映真实的情况.此外,对比不同参数得到的结果表明新参数的正确性.
2.2 构型变化
径向分布函数(RDF)描述了体系中区域密度与平均密度的比值,对于固体材料而言,通过径向分布函数可以得到不同原子之间的最近临距离,可以很好地反映不同掺杂浓度时晶格结构的变化.图2分别给出了不同势函数得到的不同掺杂浓度下,不同原子之间的第一临位距离的变化情况.

图2 不同势函数计算的各离子间最近邻距离Fig.2 Calculated nearest neighbor between different ions from different potentials
由图2可知,虽然势参数不同,但计算结果的变化趋势基本一致,验证了模拟结果的合理性.此外,图2中不同势参数得到的计算结果对比可以发现,随掺杂浓度的不断增加,阳离子与氧离子之间的最近邻距离不断变小,且Gd3+-O2-间距始终大于Ce4+-O2-间距,而O2--O2-最近邻距离却不断增大.氧空位产生后,O2-受力情况发生改变,处于氧空位临位上的O2-会产生趋向氧空位的运动,从而使整个体系中O2--O2-的最近邻距离增大,而整个过程中阳离子没有发生大的跳跃,始终在初始位置附近振荡,即阳离子之间的距离并没有发生大的变化,因此阳离子与氧离子之间的最近邻距离整体呈现减小的趋势.另外,由于在GDC中库伦力是主要的作用形式,Ce4+-O2-间的吸引作用大于Gd3+-O2-间吸引作用,因此Gd3+-O2-间距始终大于Ce4+-O2-间距.
2.3 扩散情况
一定温度下,O2-能在GDC膜中扩散是GDC膜导电的根本原因.为了研究氧离子的扩散行为,模拟了温度为1 273 K时不同掺杂浓度体系中氧离子的扩散情况,系综为NVT,时间步为1 fs,运行100万步.
均方位移(MSD)随时间的变化情况体现了模拟过程中各类原子的扩散行为,根据爱因斯坦的扩散定律[11],有

式中:D为粒子的扩散系数.当时间较长时,均方位移对时间变化曲线的斜率为6D.根据式(2)可以计算出不同势参数下氧离子的扩散系数,与实验值的对比结果如图 3,图 3中为了方便和实验值[9]对比,把CexGd1-xO2-0.5x中的x值换算成对应的Gd2O3浓度.

图3 1 273 K下不同势参数计算的O2-的扩散系数与实验值对比Fig.3 Calculated diffusion coefficients of oxygen from different potentials and experimental result
从图3可以看出,随着掺杂浓度的增大,O2-的扩散系数开始不断增大,但当掺杂浓度达到一定程度后,扩散系数却开始逐渐减小,表明了O2-的扩散行为由开始的激烈逐渐减缓,与实验现象一致.
事实上,O2-能够在GDC膜中扩散是由于氧空位的存在.初始阶段,随着掺杂浓度的增加,氧空位数量不断增多,O2-的扩散行为变得容易,即扩散系数不断增大;但同时要注意的是,氧空位显示为正电荷,掺杂离子Gd'Ce显示为负电荷,两者之间存在的库伦吸引作用会阻碍O2-的扩散,随着氧空位不断的增加,库伦吸引不断增强,对O2-扩散的阻碍作用也不断增强;以上2种效果处于竞争的状态,当掺杂浓度达到一定程度,后者的作用超过了前者,则O2-的扩散行为开始变得困难,从而扩散系数开始不断减小,正如图3所示.
此外,图3中不同参数的计算结果虽然和实验值变化趋势一致,但在具体数值上并不完全相同.实验中,当Gd2O3摩尔浓度为10%时,扩散系数最大;对于Lewis、Licia、Gotte这3组参数,当 Gd2O3摩尔浓度为8%~9%时得到的扩散系数最大,且Lewis参数提出的时间最早,其得到的模拟结果绝对值与实验值差距最大,表明该参数适用范围有限;而Hideaki的势参数,最大扩散系数对应的Gd2O3为6%~8%;新提出的势函数计算的结果与实验值变化情况较为接近,也验证了势函数的合理性.同时,不同的计算结果说明了相对于晶体构型而言,O2-的扩散行为对势参数的变化更为敏感.
3 结论
1)4套势参数及本文提出的势函数计算的GDC构型变化趋势基本一致.但提出时间较早的Lewis参数计算结果相对实验值差距较大,更适合于定性的分析,新势函数较好的符合了实验情况,验证了该势函数的正确性.
2)随着掺杂浓度的增加,GDC的晶格常数不断增加,O2--O2-的最近邻距离不断增大,而阳离子与氧离子之间的最近邻距离不断减小,且Gd3+-O2-间距始终大于Ce4+-O2-间距.
3)工作过程中,阳离子始终在初始位置发生震荡,没有发生较大的位移;O2-在GDC膜中发生了扩散.随着掺杂浓度的增加,O2-的扩散开始变得激烈,扩散系数增大,当Gd2O3摩尔浓度达到6%~8%时,掺杂离子与氧空位的相互作用会阻碍O2-扩散,使得其扩散系数逐渐降低;且不同势参数计算的扩散系数有较大差异,表明了O2-的扩散对势参数的改变更为敏感.
[1]MORALES M,ROA J J,CAPDEVILA X G,et al.Mechanical properties at the nanometer scale of GDC and YSZ used as electrolytes for solid oxide fuel cells[J].Acta Materialia,2010,58(7):2504-2509.
[2]WANG Y G,LINAN A,SARAF L V,et al.Microstructure and ionic conductivity of alternating multilayer structured Gd-doped ceria and zirconia thin films[J].Journal of Materials Science,2009,44(8):2021-2026.
[3]YE F,MORI T,OU D R,et al.Dopant type depency of domain development in rare-earth-doped ceria:an explanation by computer simulation of defect clusters[J].Solid State Ionics,2009,180(20):1127-1132.
[4]WEI X,PAN W,CHENG L F,et al.Atomistic calculation of association energy in doped ceria[J].Solid State Ionics,2009,180(1):13-17.
[5]HULL S,NORBERG S T,AHMED I,et al.Oxygen vacancy ordering within anion-deficient ceria[J].Journal of Solid State Chemistry,2009,182(10):2815-2821.
[6]SAYLE T X T,SAYLE D C.Elastic deformation in ceria nanorods via a fluorite-to-rutile phase transition[J].ASC Nano,2010,4(2):879-886.
[7]GOTTE A,SPANGBERG D,BAUDIN M.Molecular dynamics study of oxygen self-diffusion in reduced CeO2[J].Solid State Ionics,2007,178(25):1421-1427.
[8]MINERVINI L,ZACATE M O,GRIMES R W.Defect cluster formation in M2O3-doped CeO2[J].Solid State Ionics,1999,116(4):339-349.
[9]CUI Z W,SUN Y,CHEN Y J.Semi-ab initio interionic potential for gadolinia-doped ceria[J].Solid State Ionics,2011,187(1):8-18.
[10]INABA H,SAGAWA R,HAYASHI H.Molecular dynamics simulation of gadolinia-doped ceria[J].Solid State I-onics,1999,122(2):95-103.
[11]陈正隆,徐为人,汤立达.分子模拟的理论与实践[M].北京:化学工业出版社,2007:110-115.CHEN Zhenglong,XU Weiren,TANG Lida.Theory and practice of molecular dynamics simulation[M].Beijing:Chemical Industry Press,2007:110-115.
