基于EST序列的烟草cSNP发掘
2012-01-17龚达平李凤霞王卫锋刘贯山孙玉合
龚达平,王 鲁,李凤霞,王卫锋,刘贯山,孙玉合
(农业部烟草类作物质量控制重点开放实验室,中国农业科学院烟草研究所,青岛 266101)
单核苷酸多态性(single nucleotide polymerphisms,SNP)主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性。SNP在基因组中遗传稳定、分布广泛、多态性高、易实现自动化检测,被认为是最具应用潜力的第三代分子标记。除在遗传分析以及作物分子标记辅助育种中有着广阔的应用前景外,其在构建高密度遗传图谱,整合物理图谱,复杂遗传性状以及基因组进化研究等方面有着重要的应用价值[1-3]。
近几年来,新型大规模、高通量SNP发掘方法不断涌现。如DNA列阵微测序法、动态等位基因特异杂交法、寡聚核苷酸特异连接法、DNA芯片以及基质辅助激光解析电离飞行时间法等。特别是以边合成(或连接)边测序为特征的新一代测序技术的出现,如 Roche (454)GSFLX sequencer、Illumina genome analyzer (Solexa)、Applied Biosystems SOLiD sequencer、HeliScope Sequencer等[4],使基因组学和功能基因组学进入了一个低成本、大规模、高通量测序的时代。但是,普通烟草(N.tabacum)为异源四倍体(TTSS),基因组庞大,重复序列高,且T、S染色体组间存在很高的同源性。因此对烟草全基因组进行测序和组装是一项非常艰巨的工作。美国北卡州立大学仅对普通烟草基因组的基因富集区进行了测序。传统分子标记的开发耗时费力,而目前在公共数据库 GenBank中存储了超过30万条来源于不同品种的普通烟草EST序列,这为我们进行基于 EST数据的高度冗余性来发掘SNP提供了丰富的数据资源。特别是编码区 SNP(coding SNP,cSNP)位于外显子内,有可能与基因功能直接相关,因此它们在烟草的重要性状研究中具有重大的应用价值[5-6]。
目前烟草基因组及SNP发掘才刚刚开始。烟草SNP研究大多集中于对单个基因的SNP分析上,只有欧洲烟草EST测序项目在开展普通烟草EST测序工作中发掘了 74 SNPs和 38 InDels(www.ESTobacco.info)。本研究中,我们利用公共数据库中的EST序列,在无需序列质量文件的条件下保守的挖掘cSNP,并对这些cSNP位点的可信度进行评估。
1 材料与方法

2 结 果
2.1 序列组装
从GenBank中共检索到烟草EST 317 175条。这些序列所产生的cDNA文库来源的品种和组织器官以及发育时期如表1。共组装成35 504个contig和109 550个singleton(表2)。包含至少4个读长的contig共计15 429个。

表1 烟草EST数据来源的材料和组织Table 1 Cultivar and tissue of tobacco EST data
2.2 鉴定候选SNP
在包含至少4个以上reads的15 429个重叠群中,有7088个重叠群包含了冗余度为2以上的候选SNP共53 477个。不同大小的重叠群中包含候选 SNP的重叠群占该大小重叠群总数的比例如图1。图中只列出了重叠群大小为25个reads以下的分布。整体上随着contig所包含reads数量的增加,包含SNP的contig占该大小重叠群总数的比例也迅速增加。如4个reads的重叠群中包含SNP的重叠群占 13.2%,当读长数目增加到 13时,比例达到80%,而24个reads以上的重叠群中则达到94%以上,44个以上reads的重叠群全部包含SNP。
如图2,所检测到的42 420个候选SNP,其丰度也是随重叠群的增大而增加。SNP的分值也随着比对序列数目的增加而增加。平均300 bp的序列长度出现一个SNP位点。

表2 烟草EST数据不同大小重叠群的数量分布Table 2 Contigs profile of tobacco EST data

图1 不同大小的重叠群中包含候选SNP的重叠群比例Fig.1 Abundance of tobacco EST contigs containing candidate SNPs in relation to contig size.
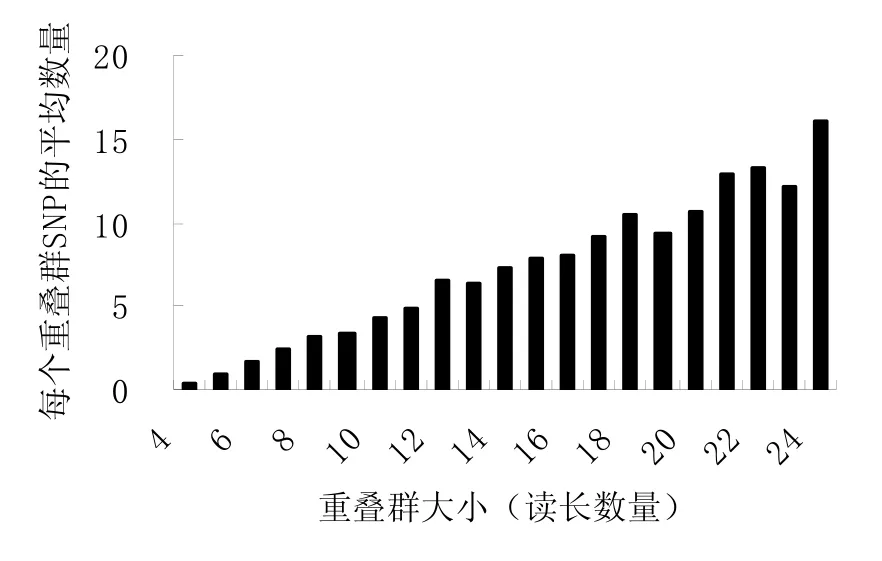
图2 不同大小重叠群包含SNP的丰度Fig.2 Abundance of candidate SNPs identified within contigs in relation to contig size for tobacco EST data
2.3 碱基变化分析
由表3可见,候选的SNP归类为转换(C/T或G/A)或颠换(C/G,A/T,C/A 或 T/G)。转换为26 582个,颠换为15 838个,转换所占的比例比颠换要大。SNP的高频转换在其他研究中也有发现,反映了甲基化后C/T的高频突变。
2.4 插入/删除分析
试验中鉴定了冗余值大于2的插入/删除(Indels)10 057个。不同长度的Indels的分步频率如表4。表4可见Indel频率随着长度的增加而降低。不管是单个还是多个碱基的 Indel,都表现出 A/T偏好(表5)。
2.5 单体型分析
除了SNP冗余度分值外,另一个SNP可信度的评价标准是通过基于对 SNP位点模式的共分离计算得到的。序列间的SNP代表了两个基因(旁系同源或直系同源)间的分化,被期望是共分离的,而在多位点上定义一个单倍型可以在一定程度上避免测序错误的随机发生。如表6和表7所示,在该重叠群中共鉴定7个候选SNP构成单体型,6个SNP位点冗余度为5,并具有较高的共分离分值和加权共分离分值,说明这些SNP位点具有较高的可信度。
比较序列来源的品种信息可以鉴定预测直系同源基因(表6)。通过随机检测100个单体型可以观察到近一半的单体型在同品系中的分离表明在单个品系中多基因的表达。尤其是在重叠群的数量和SNP的数量均比较多的情况下,这些SNP最有可能是来源于普通烟草T和S两个基因组中相似度极高的同源基因。

表3 烟草候选SNP的替换频率Table 3 Nucleotide substitution frequencies for candidate SNPs identified in tobacco EST data

表4 候选Indel数量分布Table 4 Prevalence of candidate indels identified in tobacco EST data

表5 单个和2个碱基插入删除频率Table 5 The frequency of single and dinucleotide indel sequences predicted from the alignment of tobacco EST sequences
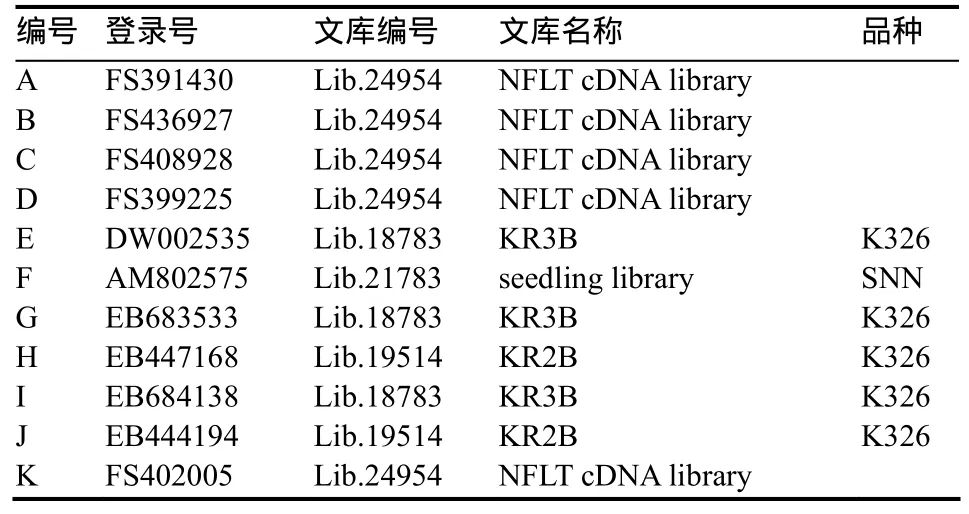
表6 重叠群包含的reads的文库和品种信息Table 6 The contig contained the sequences with ID, name and line derived from the Genbank annotation.
3 讨 论
SNP是一种可用作分子遗传分析的新型分子标记。同其他分子标记一样,其开发虽然可以自动化、规模化,但也很昂贵费力。而通过挖掘序列数据资源得到丰富的SNP是最为廉价的方法。通过筛选高质量的测序数据来发现具有生物学相关的SNP,不可避免地受到测序错误的干扰。以前主要依赖测序数据的质量文件来过滤 SNP,低质量的SNP位点可能是测序错误,而非真正的SNP位点。公共数据库中的高冗余数据往往缺乏质量文件。EST数据的冗余性允许对SNP位点的发生频率,即冗余分值,进行评估。通过检测SNP的分值大于2或更大,可以排除大多数的测序错误。虽然遗漏了一些只出现过一次的真实SNP,但数据的高冗余度允许我们从各种不同来源的数据中快速鉴定大量的SNP[9-10]。
应用这种方法我们挖掘了烟草EST数据中的SNP。目前烟草的EST数据超过30万条,通过严格的参数限制移除了54%包含少于4个读长的重叠群。大量包含4个以上读长重叠群保证了冗余度,增加了 SNP的可信度。从结果中可以看出,包含SNP的重叠群的比例和每个重叠群中 SNP的数目都随着数据量增大而增加。这里我们鉴定了42 420个候选的普通烟草SNP,这仍只是烟草遗传变异的一小部分。
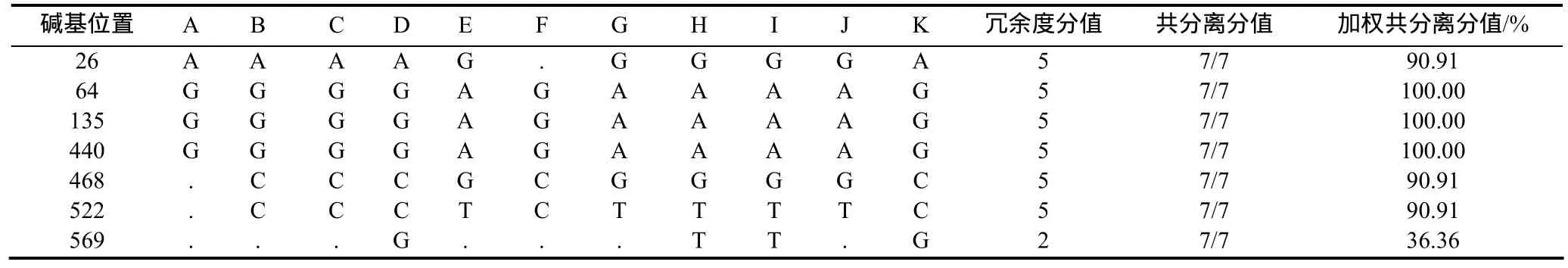
表7 重叠群中SNP统计Table 7 SNP summary in the contig
采用冗余度排除测序错误是有效的,但非随机的测序错误也会造成两次相同的序列错误。为了区分这些序列错误,需要进一步的SNP可信度衡量标准。代表两个同源基因分化的SNP会共分离形成单体型。基于一个比对中多个SNP位点的SNP模式的频率的共分离分值,允许鉴定非共分离的SNP。通过对SNP位点的数量和缺失数据的加权,能比较比对间的共分离分值。SNP分值和共分离分值一起提供评价SNP真实性的准确度,那些高冗余分值和共分离分值的SNP可能代表了真实的SNP位点。
SNP可以区分同一基因组中的复制基因(旁系同源基因)或不同品种中的直系同源基因。来自同一个品系的序列中的SNP,他们一定是由于基因复制和旁系同源基因的表达。通过随机检查100个单体型的分析表明,来自同一品种的多个基因间的SNP比例达到一半,这反映了普通烟草异源四倍体的起源,并且两个基因组高度同源。估计两个基因组间的相似程度是避免全基因组测序组装错误所必要的。因此,大量的SNP来区分烟草中的旁系同源基因有助于基因组序列的组装。
4 结 论
基于烟草EST数据的高度冗余性,317 175条普通烟草EST序列拼接成35 504个contig和109 550个singleton。其中,包含至少4个读长的contig共计15 429个,说明公共数据库中烟草EST数据足够开发大量的SNP标记。共鉴定出53 477个冗余度大于2的候选SNP,烟草中平均300 bp出现一个SNP位点。其中,SNP的转换率大于颠换,插入/删除表现出A/T偏好。结合SNP位点的冗余度和共分离分值来评价其可信度,保证了所获得的SNP位点具有较高的可信度。这些高质量SNP标记的发掘为进一步的烟草功能基因研究和分子育种奠定了基础。
[1]Gupta P K, Roy J K, Prasad M.Single nucleotide polymorphisms: a new paradigm for molecular marker technology and DNA polymorphism detection with emphasis on their use in plants[J].Curr Sci., 2001, 80:524-535.
[2]Rafalski A. Applications of single nucleotide polymorphisms in crop genetics[J].Curr Opin Plant Biol.,2002, 5(2): 94-100.
[3]Syvanen A C. Genotyping single nucleotide polymorphisms[J].Nat Rev Genet., 2001, 2(12):930-942.
[4]Voelkerding K V, Dames S A, Durtschi J D.Next Generation Sequencing: From Basic Research to Diagnostics[J].Clin Chem., 2009, 55(4):641-658..
[5]Picoult N L, Ideker T E, Pohl M G, et al.Mining SNPs from EST databases[J].Genome Res, 1999, 9(2):167-174.
[6]Lein W, Usadel B, Stitt M, et al.Large scale phenotyping of transgenic tobacco plants (Nicotiana tabacum) to identify essential leaf functions[J].Plant Biotechnol J.,2008, 6(3): 246-263.
[7]Huang X, Madan A.CAP3: A DNA Sequence Assembly Program[J].Genome Research, 1999, 9(9): 868-877.
[8]Barker G, Batley J, O’ Sullivan, et al.Redundancy based detection of sequence polymorphisms in expressed sequence tag data using autoSNP[J].Bioinformatics,2003, 19 (3): 421-422.
[9]Batley J, Barker G, O'Sullivan H, et al.Mining for Single Nucleotide Polymorphisms and Insertions/Deletions in Maize Expressed Sequence Tag Data[J].Plant Physiology, 2003, 132(1): 84-91.
[10]毛新国,汤继凤,周荣华,等.基于全长 cDNA序列的小麦 cSNP发掘[J].作物学报,2006,32(12):1836-1840.
