甜菊糖苷STV和RA的制备工艺研究
2012-01-05程新华向极钎覃大吉
程新华,向极钎,张 亮,龙 澜,覃大吉
(1.湖北省农业科学创新中心鄂西综合试验站,湖北 恩施 445000;2.恩施清江生物工程有限公司,湖北 恩施 445000)
甜叶菊(SteviarebaudianaBertoni.)为菊科,属多年生草本植物,其适应性强,在我国有广分布.甜叶菊糖苷又称甜菊苷、甜菊糖苷、甜菊糖,是从甜叶菊植物中提取,属于天然无热量高倍甜味剂.它们是一类具甜味的萜烯类配糖体,为白色粉末.甜叶菊中主要含有甜菊糖苷STV(Stevioside)、RA(Rebaudioside A)、RB(Rebaudioside B)、RC(Rebaudioside C)、RD(Rebaudioside D)和RE(Rebaudioside E)等[1-2].其中,甜菊糖苷STV成分占糖苷总量的60%~70%,其甜度为蔗糖的300倍,其次是RA,约占糖苷总量的15%~20%,其甜度为蔗糖的450倍,其他组分含量都较少[3].甜菊糖在一般加工条件下很稳定,具有非发酵性、非着色性,安全无毒,被广泛应用于食品、酿酒、医药、日用化工等行业[4].甜菊糖苷的安全性已得到国际FAO和WHO等组织的认可.我国卫生部自1985年和1990年分别批准甜菊糖苷为不限量使用的天然甜味剂和医药用甜味剂辅料,用其制成的食品饮料,长期使用不会使人发胖,特别适宜肥胖症、糖尿病、高血压、动脉硬化、龋齿病患者使用.近年来,现代药理学研究表明,甜菊糖苷还具有降低血压,促进胰岛素分泌等生物活性.因此,甜菊糖苷是一种安全的天然甜味剂,并广泛应用于食品和医药行业中,被誉为继蔗糖、甜菜糖之后的第三种天然糖源,有着广泛的应用范围和宽阔的市场前景[5-6].本文采用絮凝、吸附、脱盐、脱色和重结晶的方法分离、纯化甜菊糖苷.
1 实验部分
1.1 仪器和试剂
高效液相色谱分析仪(2695分离单元+2696二极管矩阵检测器,美国waters公司);BC-R205旋转蒸发仪(上海贝凯生物化工设备有限公司);Φ24×250 mm玻璃树脂柱;Φ35×400 mm玻璃树脂柱;层析系统(UV9091771218紫外检测器+HEP-200A恒流泵+Φ30×400 mm不锈钢层析柱+EasyChrom-1 000色谱工作站,北京慧得易科技有限责任公司);BT25S十万分之一电子天平(德国赛多利斯股份有限公司);DZKW恒温水浴锅(北京永光明医疗仪器厂);TD5A-WS台式大容量离心机(金坛市华龙实验仪器厂);CHA-SA数显气浴恒温振荡器(江苏省金坛市环宇科学仪器厂).
甜菊糖苷STV标准品(美国Sigma公司;甜菊糖苷RA成都科斯曼公司);AB-8、ADS-7、DM-130、HPD-222、HPD-X、D001、D201大孔型强碱性阴离子交换树脂(沧州宝恩公司);乙腈(色谱纯,天津市科密欧化学试剂有限公司);七水硫酸亚铁(分析纯,国药集团化学试剂有限公司);氢氧化钙(分析纯,国药集团化学试剂有限公司);乙醇(分析纯,国药集团化学试剂有限公司);甲醇(分析纯,国药集团化学试剂有限公司);正丁醇(分析纯,天津市福晨化学试剂厂);盐酸(分析纯,武汉市中天化工有限责任公司);氢氧化钠(分析纯,国药集团化学试剂有限公司).
1.2 实验方法
1.2.1 甜菊糖苷STV和RA的检测方法 甜菊糖苷STV和RA的检测方法参考国家标准GB8270-1999(食品添加剂 甜菊糖甙)和中华人民共和国药典[7].
1.2.2 甜菊糖苷提取液的制备和除杂 称取100 g干甜叶菊,按料液比1∶9加入900 mL水于50℃水浴锅中搅拌提取1.5 h,粗孔过滤,收集滤液,滤渣再按1∶8加入800 mL水于50℃水浴锅中提取1.5 h,粗孔过滤,收集滤液,滤渣再按1∶8加入800 mL水于50 ℃水浴锅中提取1.5 h,粗孔过滤,收集滤液,合并三次提取滤液.共得滤液约2 300 mL.将上述滤液平均分成两份,即每份为1150 mL.其中一份滤液加入5 g FeSO4·7H2O,边搅拌边加入CaO调节pH至9~9.5,放置2 h进行絮凝后准备过滤.由于上述两份滤液中含有大量的杂质,用滤纸抽滤的方式过滤较难进行,所以先将滤液用离心机在一定的离心条件下(4 500 r/min,10 min)进行离心后再抽滤,抽滤效果较好,即得未絮凝提取液2 200 mL,絮凝后的提取液2 100 mL.分别测定除杂率和甜菊糖苷的损失率.
1.2.2.1 除杂率的测定 分别准确量取未絮凝和絮凝后的提取液20 mL,置于蒸发皿中,水浴蒸干后,移入110℃烘箱干燥至恒重后称重,得干燥浸膏质量,按照下式计算除杂率x1:
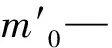
1.2.2.2 甜菊糖苷的损失率的测定 分别测定絮凝前后提取液的STV或RA的浓度,按照下式计算甜菊糖苷的损失率x2:
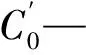
1.2.3 不同型号树脂的选型
1.2.3.1 树脂的预处理 大孔吸附树脂AB-8属于弱极性、DM-130属于中等极性、HPD-222属于中等极性、HPD-X属于中等极性.树脂的预处理:首先取一定量的树脂于烧杯中,加入纯净水,从上方将树脂碎片和其他杂质倒出,再加入纯净水直至无明显的混浊,然后将树脂中的水滤干,转移至另一个干净的烧杯中,加入95%乙醇,浸泡12 h,每2 h搅拌一次充分除去气泡.然后在树脂柱内加入适量的95%乙醇,将适量的树脂转移至树脂柱内,然后用2~4 BV的95%乙醇以2 BV/h的流速通过树脂层,直至流出液加水后无混浊现象为止.然后用纯净水冲洗树脂柱,直至流出液无乙醇为止,备用.
ADS-7属于极性树脂,树脂的预处理:首先取一定量的树脂于烧杯中,加入纯净水,从上方将树脂碎片和其他杂质倒出,再加入纯净水直至无明显的混浊,然后将树脂中的水滤干,转移至另一个干净的烧杯中,加入95%乙醇,浸泡12 h,每2 h搅拌一次充分除去气泡.然后在树脂柱内加入适量的95%乙醇,将适量的树脂转移至树脂柱内,然后用2~4 BV的95%乙醇以2 BV/h的流速通过树脂层,直至流出液加水后无混浊现象为止.然后用纯净水冲洗树脂柱,直至流出液无乙醇为止.然后再用2~3 BV的3%~5%的盐酸溶液以2 BV/h的流速通过树脂层,用水淋洗至接近中性后用2~4 BV的3%~5%的氢氧化钠溶液以2 BV/h的流速通过树脂层,用水淋洗至接近中性,备用.
D001和D201属于离子交换树脂,树脂的预处理:首先取一定量的树脂于烧杯中,加入纯净水,从上方将树脂碎片和其他杂质倒出,再加入纯净水直至无明显的混浊.然后湿法装柱,用2~3 BV的7%~8%的氯化钠溶液以2 BV/h的流速通过树脂层,并且浸泡12 h后用纯净水将树脂中的氯化钠淋洗干净.D001树脂柱首先用2~3 BV的3~5%的盐酸溶液以2 BV/h的流速通过树脂层,并且浸泡8 h,然后用水淋洗至接近中性,再用2~4 BV的3~5%的氢氧化钠溶液以2 BV/h的流速通过树脂层,并且浸泡8 h,然后用水淋洗至接近中性;D201树脂柱首先用2~4 BV的3~5%的氢氧化钠溶液以2 BV/h的流速通过树脂层,并且浸泡8 h,然后用水淋洗至接近中性后用2~3 BV的3~5%的盐酸溶液以2 BV/h的流速通过树脂层,并且浸泡8 h,然后用水淋洗至接近中性.将D001和D201树脂柱进行反洗,进一步去除预处理中释放出来的低聚物,直至反洗出水澄清为止,待再生.树脂的初次再生:D001树脂柱首先用3.5 BV的盐酸溶液以一定的流速淋洗,淋洗的时间要到达2 h,然后用脱盐水(去离子水)淋洗至基本呈中性,备用;D201树脂柱首先用3.5 BV的氢氧化钠溶液以一定的流速淋洗,淋洗的时间要到达2 h,然后用脱盐水(去离子水)淋洗至基本呈中性,备用.
1.2.3.2 大孔吸附树脂的静态吸附
1)吸附:分别取上述处理好的AB-8、ADS-7、DM-130、HPD-222、HPD-X大孔吸附树脂适量,用滤纸吸干树脂表面的水分,准确称取树脂1 g,转移至50 mL的锥形瓶中.每一种树脂取两份,分别加入上述絮凝后的提取液40 mL,将锥形瓶放置恒温振荡器中,密封,于120 r/min,25℃振荡12 h,将吸附后的溶液取样,待测,按照下式计算树脂的静态吸附量x3:
其中:x3—树脂静态吸附量,mg/g树脂;C0—吸附前提取液中甜菊糖苷STV和RA的浓度,mg/mL;C1—吸附后溶液中甜菊糖苷STV和RA的浓度,mg/mL;V1—吸附实验所取的提取液的体积,mL;m1—吸附所取的树脂的质量,g.
2)解吸:将上述的吸附饱和的树脂过滤,分离树脂,然后用适量水淋洗树脂表面吸附的提取液,然后将树脂全部转移至另外干净的50 mL的锥形瓶中,加入40 mL的70%乙醇,将锥形瓶放置恒温振荡器中,密封,于120 r/min,25℃振荡8 h,将解吸后的溶液取样,待测,按照下式计算树脂的解吸率x4:
其中:x4—树脂的解析率,%;C0—吸附前提取液中甜菊糖苷STV和RA的浓度,mg/mL;C1—吸附后溶液中甜菊糖苷STV和RA的浓度,mg/mL;V1—吸附实验所取的提取液的体积,mL;V2—解吸液的体积,mL.
1.2.3.3 大孔吸附树脂的动态吸附 将适量的提取液上大孔吸附树脂柱,上柱速度为2~3 BV/h,检测流出液中甜菊糖苷STV和RA的浓度,上柱完毕后首先用2 BV的脱盐水以3 BV/h进行除杂,然后再用一定浓度的乙醇溶液洗脱,洗脱速度为2~3 BV/h,收集洗脱液,浓缩回收乙醇,得甜菊糖苷待脱盐液.取适量的洗脱液干燥,检测其中STV和RA的纯度.
1.2.3.4 脱盐、脱色 将D001和D201树脂串联,甜菊糖苷待脱盐溶液上柱,上柱速度为2~3 BV/h,检测流出液中甜菊糖苷STV和RA的浓度,上柱完毕后首先用2 BV的脱盐水以3 BV/h进行除杂,然后用一定浓度的乙醇溶液分别洗脱D001和D201,并且分别收集D001和D201树脂柱的洗脱液,将D001和D201树脂柱的洗脱液干燥后检测STV和RA的纯度.
1.2.3.5 重结晶纯化甜菊糖苷STV和RA 采用不同的溶剂,如甲醇、乙醇、正丁醇及其溶液在适当的温度下溶解,然后在室温以及冰箱中冷藏结晶,将晶体洗涤后干燥,检测晶体中的STV和RA纯度.
2 结果与讨论
2.1 甜菊糖苷提取液的除杂
称取干甜叶菊100 g,按上述方法提取后得提取液,然后将提取液平均分为两份,其中一份未絮凝,另外一份用FeSO4·7H2O絮凝,比较絮凝前后的除杂效果和甜菊糖苷的损失率.甜菊糖苷STV和RA的标准品HPLC检测图谱见图1,絮凝前的STV和RA的HPLC检测图谱见图2,絮凝后的STV和RA的HPLC检测图谱见图3.

图1 STV和RA标准品的HPLC色谱图 图2 絮凝前甜叶菊提取液的HPLC色谱图
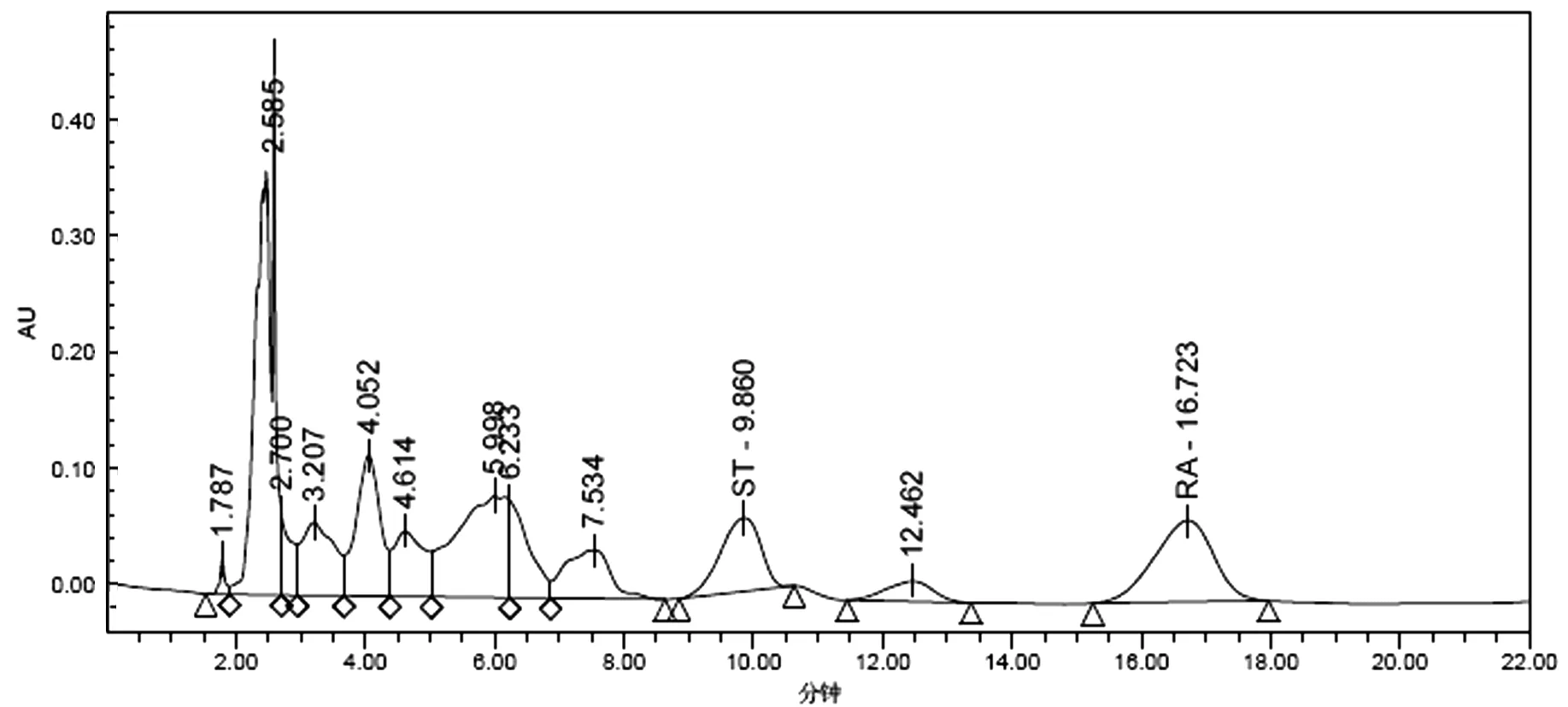
图3 絮凝后甜叶菊提取液的HPLC色谱图
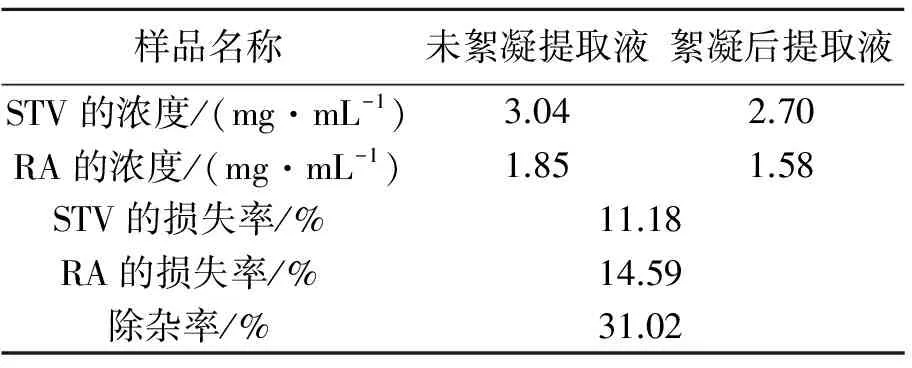
表1 絮凝前后的除杂效果和甜菊糖苷的损失率
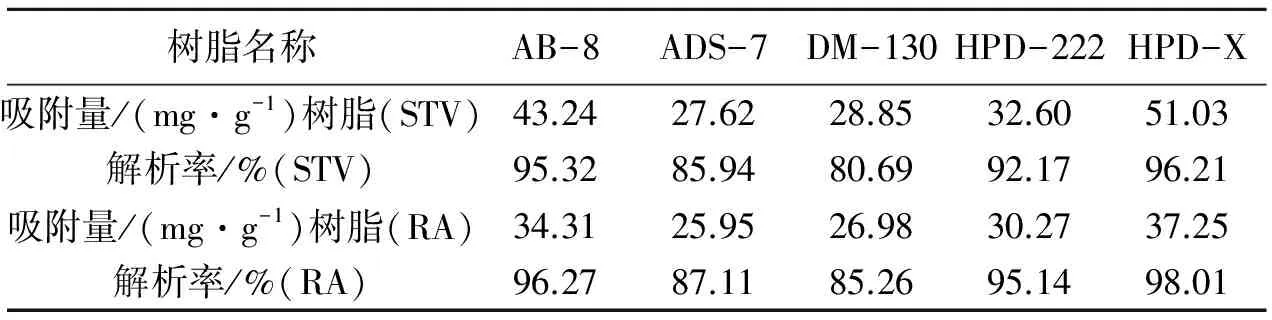
表2 不同大孔吸附树脂的静态吸附量和解吸率
从图2和图3可知,絮凝后的图谱中,STV和RA的峰面积小于絮凝前的峰面积,说明絮凝会降低STV和RA的浓度,会造成STV和RA损失.
絮凝前后的除杂效果和甜菊糖苷的损失率见表1.从表1可知,用硫酸亚铁絮凝后,甜菊糖苷STV和RA会有一定的损失,是由于在化学絮凝时,絮凝剂间会发生化学反应生成胶核,由于胶核带正电荷使提取液中的带负电的杂质聚沉,而且由于胶体的架桥作用,也会使提取液中的其他的大分子物质沉降.当胶核过量时,同时吸附甜菊糖苷分子中的羟基上的孤对电子,使甜菊糖苷聚沉,所以在加入絮凝剂量大时,甜菊糖苷的损失会较大.由于絮凝后溶液的澄清度明显大于未絮凝的溶液,除杂率达到30%以上,除杂效果较好.所以即使絮凝会加大甜菊糖苷的损失,但是由于絮凝可以出去提取液中的一些杂质,减轻了后续吸附和脱盐、脱色负荷,絮凝工艺是可取的除杂工序.
2.2 大孔吸附树脂的静态吸附
取50 g干甜叶菊,按上述步骤提取和絮凝,制备得絮凝后的提取液.分别称取1 g预处理好的AB-8、ADS-7、DM-130、HPD-222、HPD-X树脂,按静态吸附实验方法进行大孔吸附树脂的静态吸附和解吸.静态吸附实验的吸附量和解吸率见表2.
从表2可以看出,HPD-X大孔吸附树脂的吸附量和解吸率在上述树脂中是最高的,适合吸附甜菊糖苷.ADS-7树脂的脱色效果最好,在吸附的过程中,吸附后的溶液颜色最浅,树脂颜色最深,与文献[8]的结果一致,但是ADS-7树脂的吸附量和解吸率都不高,不适合吸附甜菊糖苷.DM-130和HPD-222树脂吸附量都不高,也不适合吸附甜菊糖苷.HPD-X大孔吸附树脂是一种新型的接枝聚合物,利用聚苯乙烯共聚物中的悬挂双键,将极性基团负载到树脂骨架上,在增加树脂表面极性、提高吸附选择性的同时,避免了传统极性单体与苯乙烯共聚不均匀造成的塌孔,使树脂保持了高的比表面积,高吸附容量.
2.3 大孔吸附树脂的动态吸附
取500 g干甜叶菊,按上述步骤提取和絮凝,制备得絮凝后的提取液.将提取液平均分为五份.分别称取100 g预处理好的AB-8、ADS-7、DM-130、HPD-222、HPD-X树脂,加入树脂柱中.将提取液以2~3 BV/h进行上柱,刚开始每50 mL取样一次,当流出液达到1 000 mL时,每20 mL取样一次,直至出现漏点即流出液含有STV和RA.然后用2 BV的脱盐水以2~3 BV/h进行除杂,然后用4 BV的70%乙醇以2~3 BV/h进行洗脱,收集洗脱液,将洗脱液回收乙醇后得无醇待脱盐液.取适量的洗脱液干燥,检测其中STV和RA的纯度.动态吸附实验结果见表3.
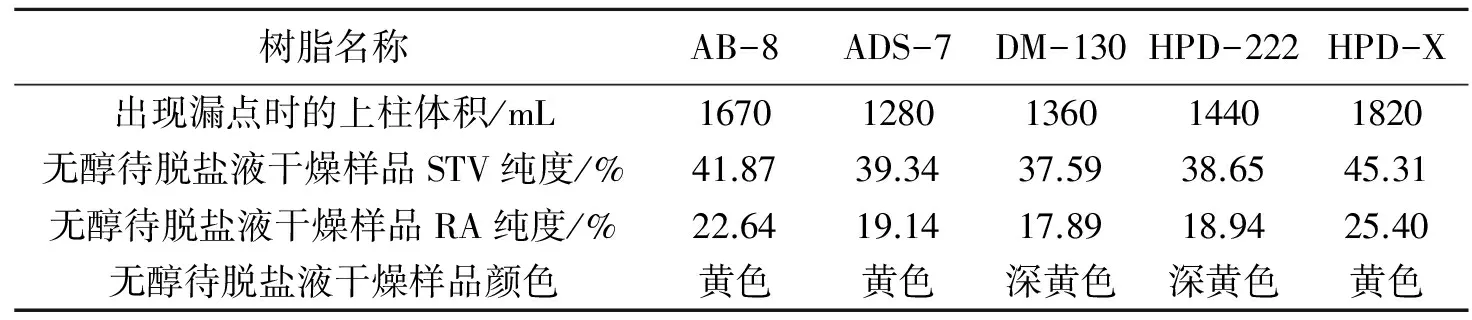
表3 不同大孔吸附树脂的动态吸附实验结果
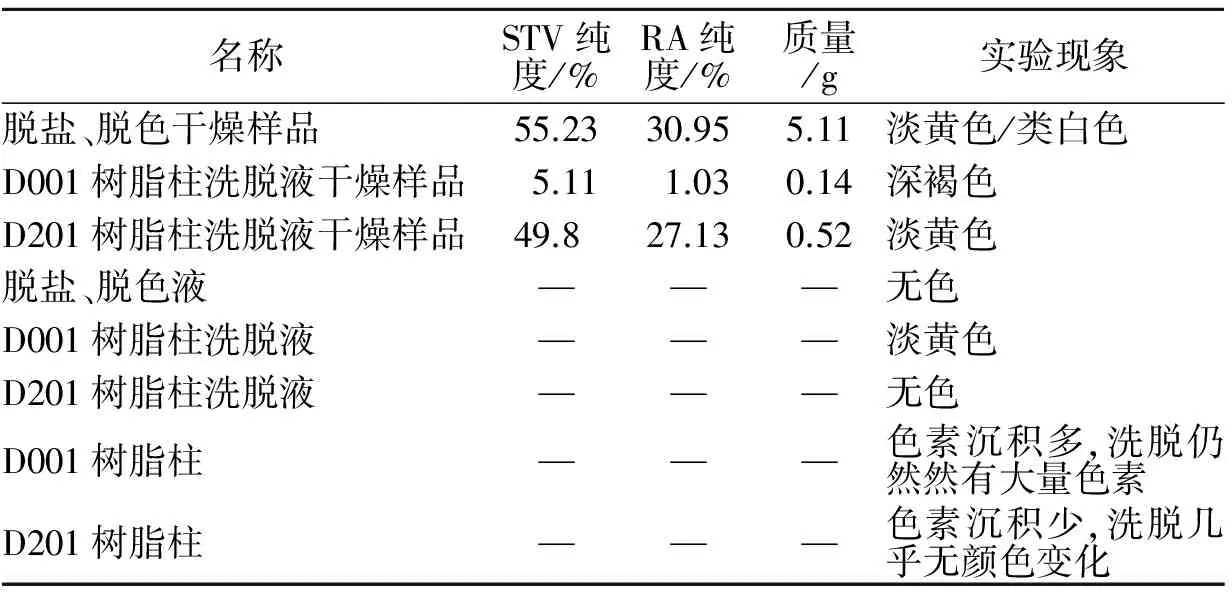
表4 脱盐和脱色实验结果
从表3可知,HPD-X出现漏点时的流出液的体积为1 820 mL,大于其他类型的大孔吸附树脂,即HPD-X的动态吸附量大于其他类型的树脂,与静态吸附实验结果一致.从待脱盐液干燥样品的纯度看,HPD-X树脂初步除杂后的样品中的STV和RA的纯度达到45.31%和26.40%,均高于其他树脂,说明HPD-X适合初步纯化甜菊糖苷提取液.
2.4 脱盐、脱色
分别称取100 g经过预处理并且再生为H-型和OH-型的D001和D201离子交换树脂,湿法装柱.将二者串联并且待上柱液上柱时先通过阳离子交换树脂D001再通过阴离子交换树脂D201进行脱盐、脱色.将2.3节中经过HPD-X初步纯化的无醇待脱盐液以2~3 BV/h速度进行上柱,收集流出液I.上柱完毕后先用2 BV的脱盐水以2~3 BV/h速度进行淋洗,收集流出液II,将流出液I和II合并,真空干燥得脱盐、脱色干燥样品.然后将D001和D201树脂柱用70%乙醇分别洗脱,70%乙醇用量各为4 BV,洗脱速度为2~3 BV/h.分别收集D001树脂柱洗脱液和D201树脂柱洗脱液,减压浓缩,干燥得D001树脂柱洗脱液干燥样品和D201树脂柱洗脱液干燥样品.脱盐、脱色实验结果见表4.
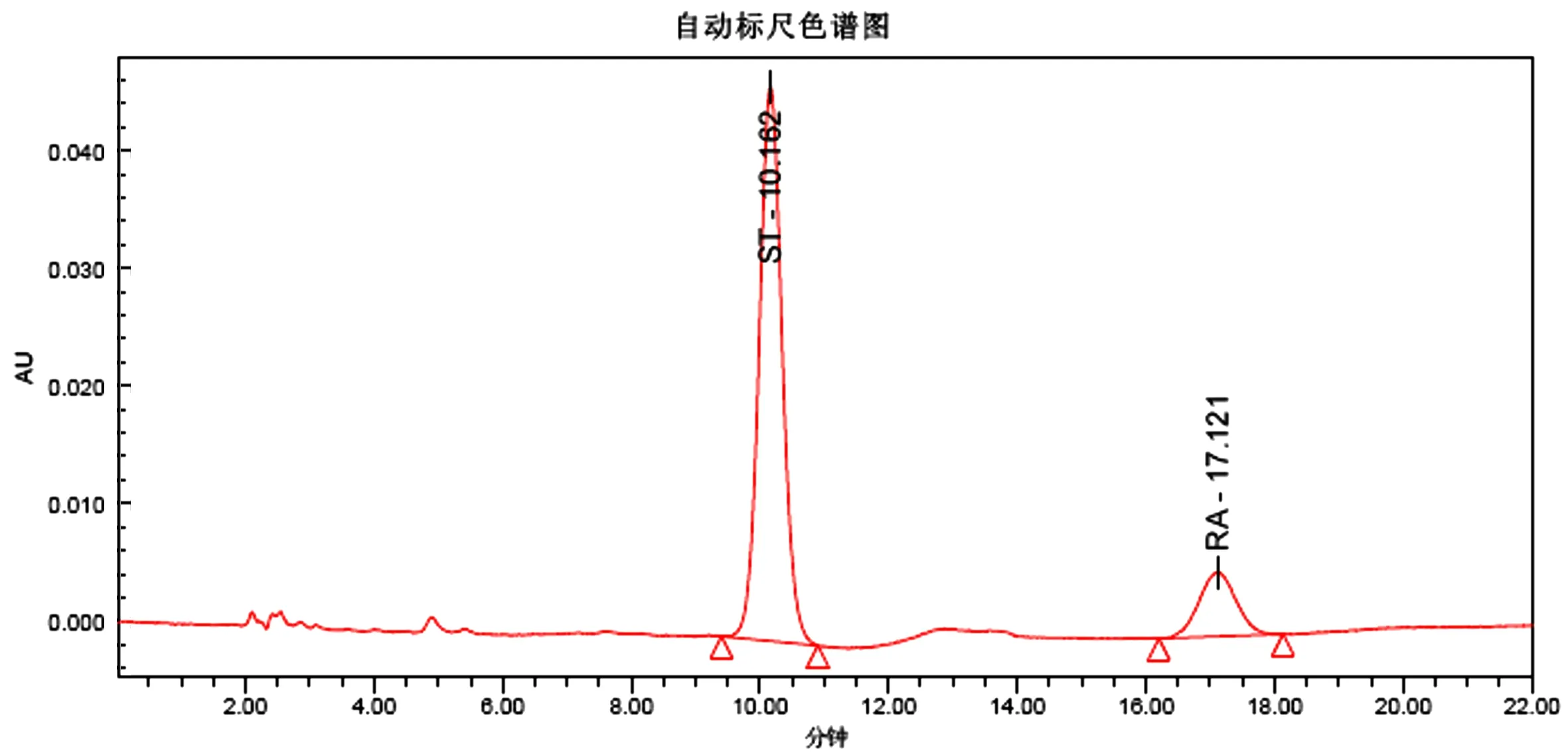
图4 高纯度甜菊糖苷样品的HPLC色谱图
从表4可知,用D001和D201串联可以对甜菊糖苷溶液进行脱盐和脱色,通过脱盐和脱色工序可以提高样品的纯度,STV和RA的纯度达到85%以上而且甜菊糖苷样品的色度也有很大的改善.D001树脂可以吸附大量的色素,无醇待脱盐液干燥样品颜色为黄色,通过脱盐、脱色工序后样品的颜色为淡黄色或白色,说明脱色效果达到一定的效果,而且D001树脂柱用70%乙醇洗脱后的的干燥样品的质量仅为0.14 g,说明D001树脂对甜菊糖苷STV和RA几乎不吸附或者吸附量很少.但是,D201树脂柱用70%乙醇洗脱后的的干燥样品的质量达到0.52 g,说明D201树脂对甜菊糖苷STV和RA有一定的吸附,需要将D201树脂柱中吸附的甜菊糖苷洗脱下来.所以在实际的脱盐、脱色过程中,D001树脂柱不需要用70%乙醇进行洗脱,直接用酸、碱液再生即可,而D201树脂柱则需要用一定量的70%乙醇进行洗脱,将流出液和洗脱液合并,减压回收溶剂,干燥制得甜菊糖苷脱盐、脱色样品.
2.5 重结晶纯化甜菊糖苷
取2.4节制备脱盐、脱色干燥样品0.1 g,加入0.4 mL甲醇于50℃水浴中溶解,然后放置0~4℃冷却结晶10 h,分离晶体和母液,晶体用1 mL的0~4℃甲醇洗涤两次,将晶体干燥,得高纯度的甜菊糖苷样品.用HPLC测得高纯度的甜菊糖苷样品中的STV纯度达85.04%,RA的纯度达13.27%,甜菊糖苷的总纯度达98%以上,重结晶收率达85%以上.高纯度甜菊糖苷样品HPLC检测图谱见图4.
3 结论
通过上述的实验可知,首先将甜叶菊用水提取出来,然后经过絮凝、吸附、脱盐、脱色和重结晶工序可以制备高纯度的甜菊糖苷样品,其STV和RA的总纯度可以达到98%以上.甜菊糖苷提取液用硫酸亚铁盐进行絮凝,可知除去提取液中的不少杂质,减少后续树脂的处理负荷;大孔吸附树脂HPD-X吸附量大,而且解吸率高,适合甜菊糖苷提取液的初步纯化,树脂的吸附量和解吸率能够满足工艺需要,而且树脂通过酸、碱再生可以重复使用,再生后的树脂的吸附量接近新的树脂;将D001和D201树脂串联可以对甜菊糖苷进行脱盐和脱色,改善甜菊糖苷的品质,由于D001树脂对甜菊糖苷的吸附量小,不需要用乙醇进行洗脱,直接再生即可,而D201树脂对甜菊糖苷有一定吸附作用,需要用70%乙醇进行洗脱以提高甜菊糖苷的产率,离子交换树脂通过酸、碱再生可以重复使用;重结晶可以进一步提纯甜菊糖苷样品,重结晶收率达85%以上,STV和RA的总纯度达98%以上.该工艺操作简单,产品能满足市场需求,适合工业化生产.
[1] 孙传范,李进伟.甜菊糖苷研究进展[J].食品科学,2010,31(9):38-340.
[2] 马磊,石岩.甜叶菊的综合开发利用[J].中国糖料,2009(1):68-72.
[3] 于聪敏,石岩.甜菊糖甙的测定方法[J].中国糖料,2009(1):65-67.
[4] 李培,杨瑞金,华霄,等.重结晶法分离甜菊糖的工艺研究[J].食品与机械,2010,26(1):160-163.
[5] 邸多隆,黄新异,陈振斌.从甜叶菊中制备甜菊糖苷的工艺方法[P].中国专利:CN 102060891 A,2011-05-18.
[6] 王贵明,董振红,郝再彬.甜叶菊糖苷的应用和安全性的研究进展[J].中国食品添加剂,2007(6):65-69.
[7] 国家药典委员会.中华人民共和国药典[S].北京:化学工业出版社,2000:771-772.
[8] 施荣富,史作清,王春红,等.吸附树脂的色层吸附及其在甜菊甙分离纯化中的应用[J].离子交换与吸附,2001,17(1):23-30.
