Complete mitochondrial genome of the laced fritillary Argyreus hyperbius (Lepidoptera: Nymphalidae)
2011-12-25WANGXiaoCanSUNXiaoYanSUNQianQianZHANGDaXiuHUJingYANGQunHAOJiaSheng
WANG Xiao-Can, SUN Xiao-Yan, SUN Qian-Qian, ZHANG Da-Xiu , HU Jing, YANG Qun,*, HAO Jia-Sheng,,*
(1. College of Life Science, Anhui Normal University, Wuhu 241000, China; 2. LPS, Institute of Geology and Paleontology, the Chinese Academy of Sciences, Nanjing 210008, China)
Complete mitochondrial genome of the laced fritillaryArgyreus hyperbius(Lepidoptera: Nymphalidae)
WANG Xiao-Can1, SUN Xiao-Yan2, SUN Qian-Qian1, ZHANG Da-Xiu1, HU Jing1, YANG Qun2,*, HAO Jia-Sheng1,2,*
(1. College of Life Science, Anhui Normal University, Wuhu 241000, China; 2. LPS, Institute of Geology and Paleontology, the Chinese Academy of Sciences, Nanjing 210008, China)
We investigated the complete mitochondrial genome (mitogenome) ofArgyreus hyperbius. The 15 156 bp long genome harbored the gene content (13 protein coding genes, 22 tRNA genes, 2 rRNA genes and an A+T-rich region) and the gene arrangement was identical to all known lepidopteran mitogenomes. Mitogenome sequence nucleotide organization and codon usage analyses showed that the genome had a strong A+T bias, accounting for A+T content of 80.8%, with a small negative AT skew (−0.019). Eleven intergenic spacers totaling 96 bp, and 14 overlapping regions totaling 34 bp were scattered throughout the whole genome. As has been observed in other lepidopteran species, 12 of the 13 protein-coding genes (PCGs) were initiated by ATN codons, while the COI gene was tentatively designated by the CGA codon. A total of 11 PCGs harbored the complete termination codon TAA, while the COI and COII genes ended at a single T residue. All of the 22 tRNA genes showed typical clover structures except that the tRNASer(AGN)lacks the dihydrouridine (DHU) stem which is replaced by a simple loop. The intergenic spacer sequence between the tRNASer(AGN)and ND1 also contained the ATACTAA motif, which is conserved in all other lepidopterans as well. Additionally, the 349 bp A+T-rich region was not comprised of large tandem repetitive sequences, but harbored a few structures common to other lepidopteran insects, such as the motif ATAGA followed by a 20 bp poly-T stretch, a microsatellite-like (AT)9element preceded by the ATTTA motif, and a 5 bp poly-A site present immediately upstream of tRNAMet. The mitochondrial genomic sequence features found in this study not only contribute to genetic diversity information of the group, but also are useful in future studies of the endangered nymphalid butterfly in population genetic dynamics, species conservation, phylogeography and evolution.
Argyreus hyperbius; Nymphalidae; Lepidoptera; Mitochondrial genome
The laced fritillary,Argyreus hyperbiusLinnaeus, is an oriental nymphalid butterfly species distributed in areas of south-east Asia, India, and north-east Africa. In recent decades, mainly owing to habitat destruction, numerous local populations have shown a sharp decline, and thus this species is considered endangered in some countries including China. Known as the “flying flower”,A. hyperbiuswas once wide-spread but is now rarely found in any large cities, such as Nanjing (Wu, 2008). To date, however, this once widely distributed species has received little attention. Detailed research focusing on aspects such as population genetic divergence, phylogeography and other relevant areas are required; thus, our study was conducted to assist in the protection and better understanding of this butterfly species.
Animal mitochondrial genomes are generally a circular molecule, ranging from 15−20 kb in size, and with a few exceptions, they all encode 37 genes: 13 protein-coding genes (PCGs), 2 ribosomal RNA genes (lrRNA and srRNA), and 22 transfer RNA genes and non-coding control elements regulating the transcription and replication of the mitochondrial genome (Taanman, 1999). Maternally inherited mtDNA is simple and stable in structure. These genes are predominantly encoded on both strands and are compactly arranged, with coding segments separated by none or only very short (a few base pairs) non-coding spacers, and in rare cases, a few genes overlap. Therefore, mitochondrial genes or genomes have been used as potential tools in studies of phylogenetics, phylogeography, phylogenetic chronology, and molecular diagnostics (Nardi et al, 2005; Simonsen et al, 2006) especially with the aid of PCR methodologies (Kocher et al, 1989; Yamauchi et al, 2004).
Within the Lepidoptera order, the butterflies (Rhopalocera) account for nearly 16 000 species, and its largest subgroup (Nymphalidae) contain approximately 5000 species (DeVries, 2001). Despite this large taxonomic diversity, information about the nymphalid butterfly mitogenome is still limited, and to the best of our knowledge, only a few complete or nearly complete mitogenomes of nymphalid species are currently available on GenBank (Tab.1). Thus, newly added mitogenome sequences of nymphalid species can providefurther insights into their diversity and evolution. In this study, we sequenced the entire mitogenome of the nymphalid butterflyArgyreus hyperbiusand analyzed its nucleotide organization and major characteristics compared with those of other butterfly species to increase of understanding of mitogenomes and phylogenies of correlative butterflies.
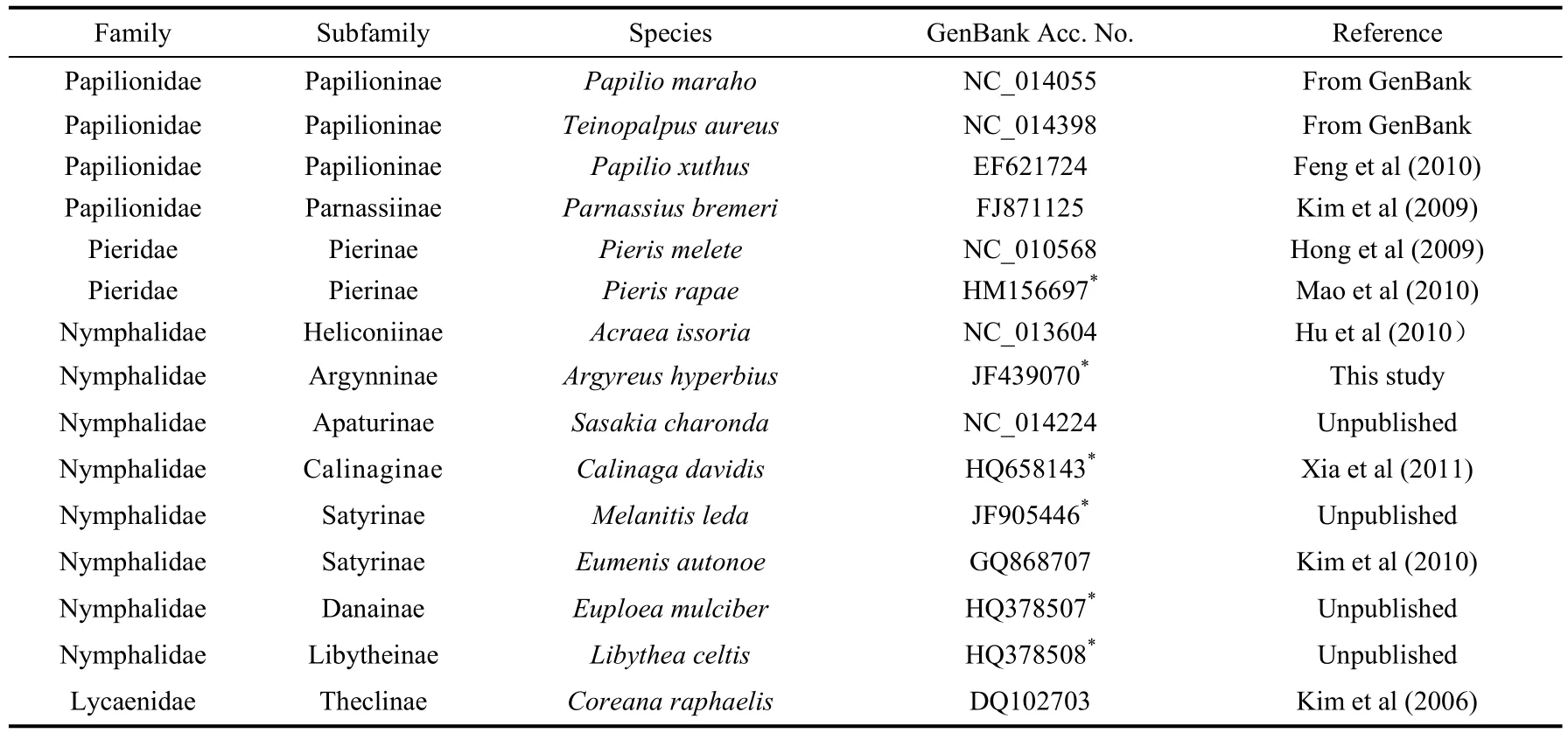
Tab. 1 Mitochondrial genomes employed in this study
1 Materials and Methods
1.1 Sample and DNA extraction
AdultA. hyperbiusindividuals were collected on Huangshan Mountain in Anhui Province, China, on August 2006 (specimen voucher ZWX09). After collection, the fresh materials were preserved in 100% ethanol immediately and stored in a −20 °C refrigerator before genomic DNA extraction.
Whole genomic DNA was extracted and purified by the modified glass powder method, whereby rice-sharp thorax muscle taken and put into one 10 mL Eppendorf tube, washed twice with ddH2O, soaked for about 2−3 h, and then incubated with 500 μL DNA liquid (5 mmol/L of NaCl, 0.5% SDS, 15 mmol/L of EDTA, 10 mmol/L of Tris-HCl, pH 7.6) and 40 μL of Proteinase-K (20 mg/ml), After this, the muscle was bathed at 55 °C for 10−12 h and centrifuged at 4 000 rpm for 2 min. The liquid supernatant was diverted into a new 10 mL Eppendorf tube, to which 500 μL of 8 mol/L GuSCN and 40 μL of 50% clean glass liquid mixture was added and the solution was then bathed at 37 ℃ for 1−2 h, rocked for 1 h, and centrifuged at 4 000 r/min for 1 min. The supernatant was then removed and the sediments were twice cleaned with 75% alcohol and once with acetone, and dried thoroughly in a vacuum dryer at 45 °C. Then 60 μL of TE (10 mmol/L Tris-Cl, 1 mmol/L EDTA, pH 8.0) was added into the Eppendorf tube with powder, and bathed at 56 °C for 30 min, then finally speed up slowly till 4 000 r/min and centrifuged for 1 min. The supernatant containing the genomic DNA was then transferred into a clean 1.5 mL Eppendorf tube and preserved at −20 °C till use (Hao et al, 2007).
1.2 PCR amplification and sequencing
Some universal primers for short fragment amplifications of 12S rRNA, COI, Cyt b genes were used for PCR (Simon et al, 1994; Simons & Weller, 2001). Long primers and some short ones including COIII and ND5 were designed by the multiple sequence alignments of all the available complete lepidopteran mitochondrial genomes (Tab. 1) using ClustalX1.8 (Thompson et al, 1997) and Primer Premier 5.0 software (Singh et al, 1998).
Long PCRs were performed using TaKaRa LA Taq polymerase with the following cycling parameters: initial denaturation for 5 min at 95 °C, followed by 30 cycles at 95 °C for 50 s, 50 °C for 50 s, 68 °C for 2 min and 30 s; and a final extension step of 68 °C for 10 min. Short fragments were amplified with TaKaRa Taq polymerase: initial denaturation for 5 min at 94 °C, followed by 35 cycles at 94 °C for 1 min, 45−53 °C for 1 min, 72 °C for 2 min, and a final extension step of 72 °C for 10 min. The PCR products were detected via electrophoresis in 1.2% agarose gel, purified using the 3S Spin PCR Product Purification Kit and sequenced directly with an ABI-3730 automatic DNA sequencer.
1.3 Sequence analysis
The determined sequences were checked firstly with the NCBI Internet BLAST search function. Raw sequence files were proof read and assembled in BioEdit version 7.0 (Hall, 1999) as well as ClustalX 1.8 (Thompson et al, 1997). Transfer RNA gene analysis was conducted using tRNAscan-SE software v.1.21 (Lowe & Eddy, 1997). Putative tRNA genes not found by tRNAscan-SE were confirmed by sequence comparison betweenA. hyperbiusand other lepidopterans. Both PCGs and ribosomal RNA genes were identified by ClustalX1.8 software, and the PCGs nucleotide sequences were translated on the basis of the Invertebrate Mitochondrial Genetic Code. Nucleotide composition skewness (AT skew=(A−T)/(A+T), GC skew=(G−C)/(G+C) (Irwin et al, 1991)) and codon usage were calculated in MEGA 4.0 software (Kumar et al, 2004). TheA. hyperbiusmitogenome sequence data were deposited into the GenBank database under the accession number JF439070.
2 Results
2.1 Genome organization
The mitogenome ofA. hyperbiuswas 15 156 bp in length (Fig. 1) and encoded 37 genes totally 13 PCGs (ATP6, ATP8, COI-III, ND1-6, ND4L, Cyt b), 2 ribosomal RNA genes for small and large subunits (srRNA and lrRNA), and 22 transfer RNA genes) and a non-coding A+T-rich region (the control region) (Tab. 2). Among these, 14 genes were encoded on the N strand, including 4 PCGs (ND1, ND4, ND4L, ND5), 2 ribosomal RNA genes for small and large subunits, and 8 transfer RNA genes (tRNAGln, tRNACys, tRNATyr, tRNAPhe, tRNAHis, tRNAPro, tRNALeu(CUN), tRNAVal). The remaining 22 genes and A+T-rich region were encoded on the J strand. Eleven intergenic spacers totaling 96 bp, and 14 overlapped regions totaling 34 bp were scattered throughout the whole genome.
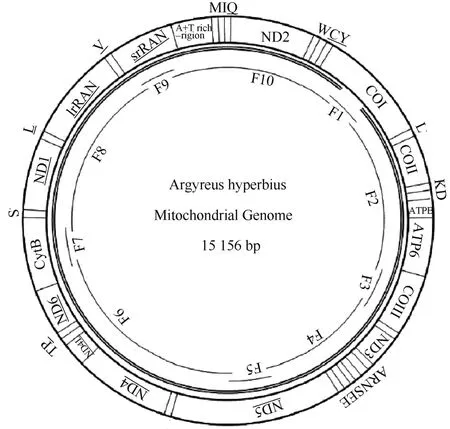
Fig. 1 Circular map of the Argyreus hyperbius mitochondrial genome
2.2 PCGs, tRNA and rRNA genes, A+T-rich region
Twelve of the 13 PCGs were initiated by ATN codons, while the COI gene was tentatively designated by the CGA codon; eleven PCGs harbored the complete termination codon TAA, while the COI and COII genes ended at a single T residue.
Results showedA. hyperbiusharbored the typical set of 22 tRNA genes ranging from 61 to 71 bp in size. All the predicted secondary structures of theA. hyperbiustRNAs are shown in Fig. 2. Some 22 tRNA genes showed typical clover structures except that the tRNASer(AGN)lacked the dihydrouridine (DHU) stem, which was replaced by a simple loop. Seventeen tRNA genes has a total of 26 pair mismatches in their stems, among which, seven were in the DHU stems, nine in the amino acid acceptor stems, one in the TΨC stem, and nine in the anticodon stems, respectively.
Based on the mitogenomes of the other insects, two rRNA genes (lrRNA and srRNA) were present inA. hyperbius. The 1 330 bp lrRNA and 778 bp srRNA were located between tRNALeu(CUN) and tRNAVal, and between tRNAValand the A+T-rich region, respectively.
The 349 bp A+T-rich region was not comprised of large tandem repetitive sequences, but harbored a few structures common to other lepidopteran insects, such as motif ATAGA followed by a 20 bp poly-T stretch, a microsatellite-like (AT)9element preceded by the ATTTA motif, and a 5 bp poly-A site present immediately upstream of tRNAMet.
2.3 Sequence variation and codon usage
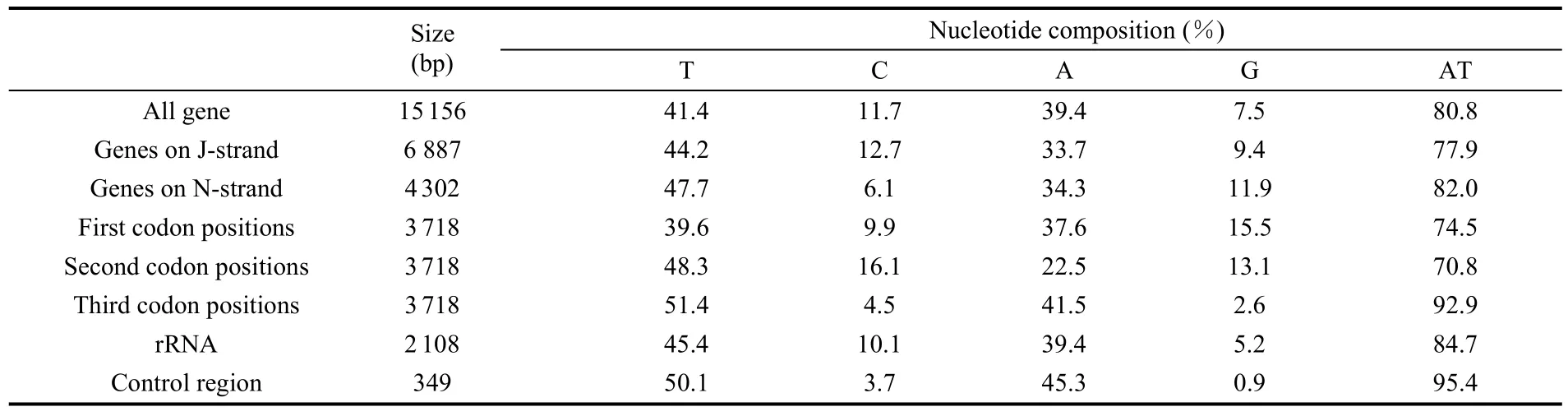
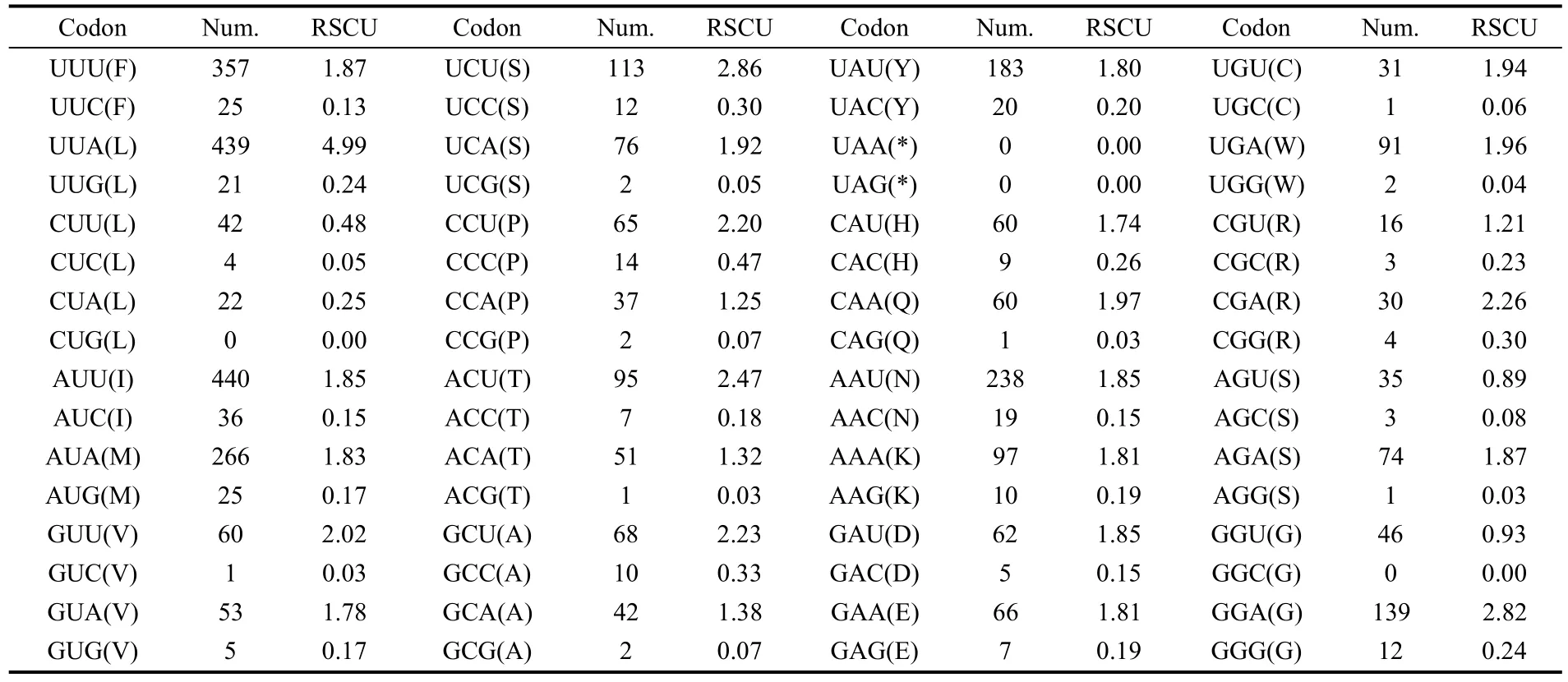
The A+T content of theA. hyperbiuswas 80.8%, and the whole mitogenome showed obvious A+T bias (Tab. 3). The relative synonymous codon usage (RSCU) in theA. hyperbiusmitochondrial PCGs was investigated and the results are summarized in Tab. 4. The four most frequently used codons were TTA (leucine, Leu), ATT (isoleucine, Ile), TTT (phenylalanine, Phe), and ATA (methionine, Met), accounting for 40.4% of all the codons in theA. hyperbiusmitogenome. These four codons were composed of A or T nucleotides, indicating their biased usage. The total number of non-stop codons (CDs) of theA. hyperbiusmitochondrial PCGs was 3 718. Among these amino acid codons, the Leu (14.20%), Ile (12.80%), Phe (10.27%), and Ser (8.50%) were the most frequently used.
3 Discussion
3.1 Genome organization
The size of the mitogenome was congruent with the sizes of other known lepidopteran mitogenomes, ranging from 15 122 bp inMelanitis leda(unpublished, GenBank accession number JF905446) to 16 094 bp inPapilio maraho(unpublished, NC_014055). The gene content of theA. hyperbiusmitogenome was the same as the typical animal mitogenome, and the gene order and orientation were identical to the already determined lepidopteran mitogenomes. Compared with other lepidopterans, however, theA. hyperbiusmitogenome was relatively more compacted, with a total of only 96 bp intergenic spacers ranging from 2−52 bp in length. Additionally, a total of 34 bp overlapped regions were scattered throughout the whole genome. Its tRNA cluster existing ahead of NADH dehydrogenase subunit 2 (ND2) was arranged in M-I-Q order, which means the tRNAMet(M) was followed by tRNAIle(I) and tRNAGln(Q), which was similar to lycaenidCoreana raphaelis(Kim et al, 2006) and the noctuidOchrogaster lunifer(Salvato et al, 2008). As far as we know, all determined lepidopteran genomes, including that ofA. hyperbius, share the same order of gene arrangement but differ from that of hypothesized ancestral insects. This confirms the suggestion proposed by Boore et al (1998) that the Lepidoptera may have diverged from other insect orders for a certain period of time, forming an independent evolutionary lineage.
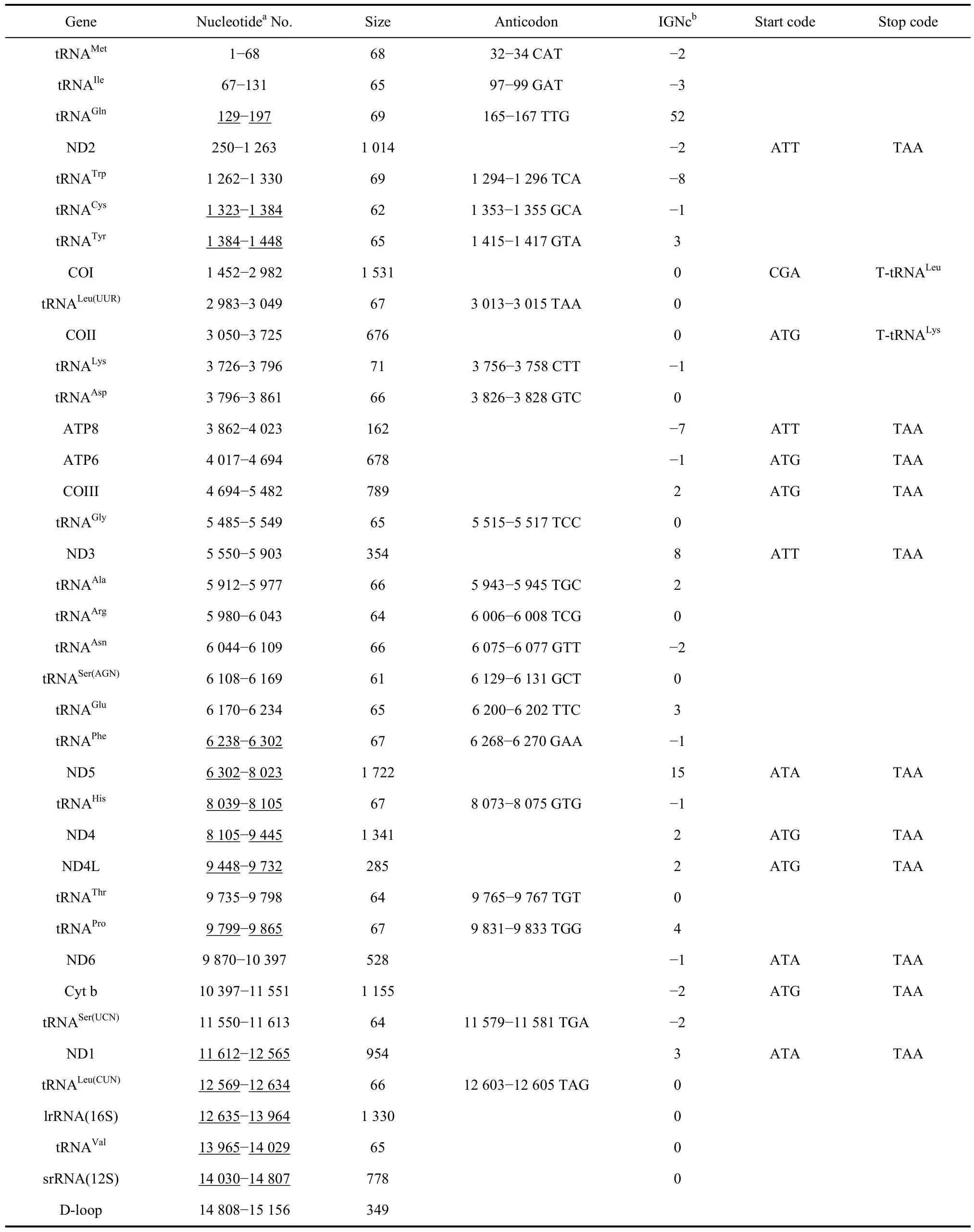
Tab. 2 Organization of the Argyreus hyperbius mitochondrial genome
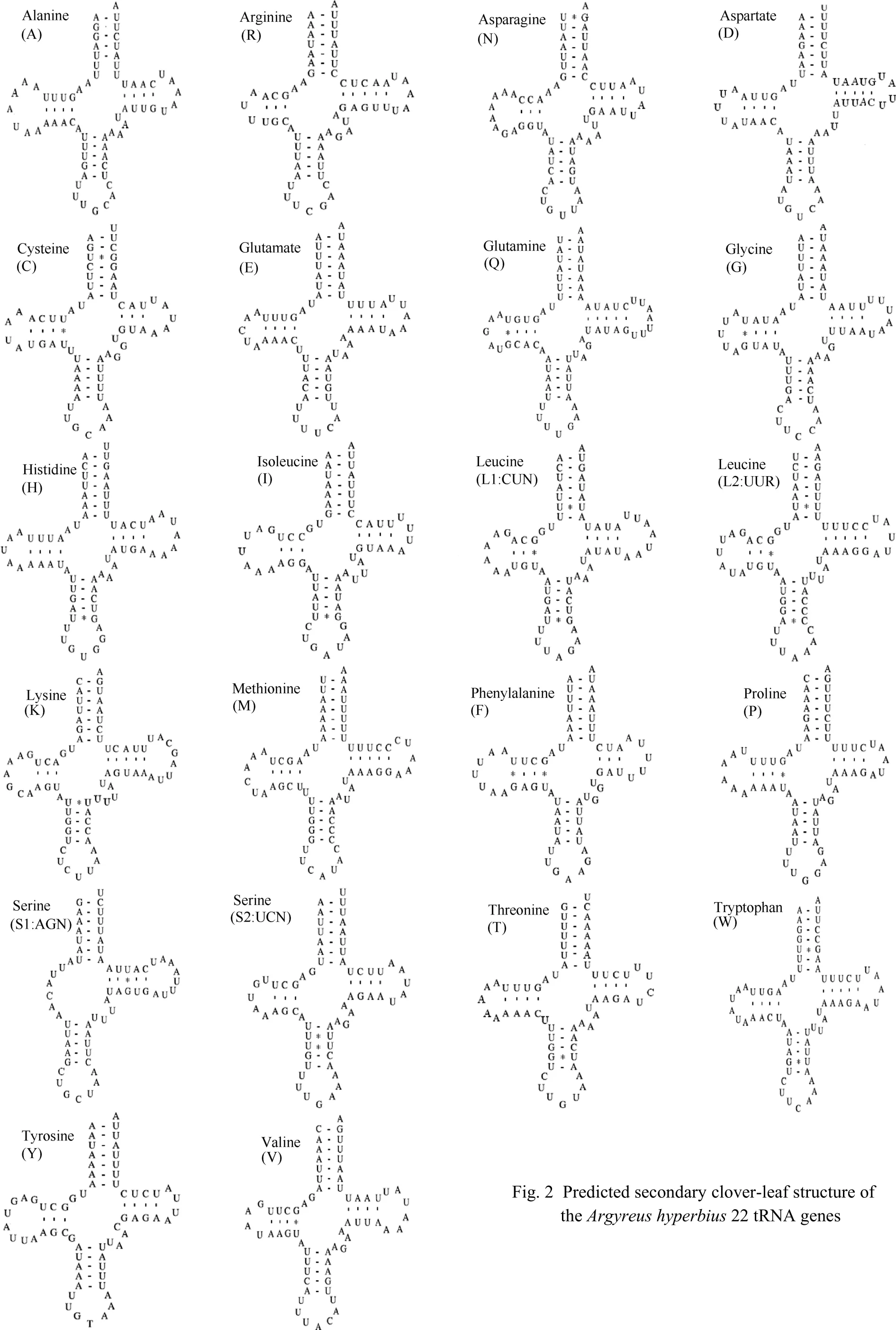
Fig. 2 Predicted secondary clover-leaf structure of the Argyreus hyperbius 22 tRNA genes
Tab. 3 Nucleotide composition and skewness in different regions of the Argyreus hyperbius mitogenome
Tab. 4 The codon number and RSCU in the Argyreus hyperbius mitochondrial PCGs
3.2 Protein-coding genes
All protein-coding sequences except COI gene use standard ATN start codon inA. hyperbius(Tab. 2). Three PCGs (ND5, ND1 and ND6) were initiated by ATA (Met); six PCGs (COII, ATP6, COIII, ND4, ND4L and Cyt b) were initiated by ATG (Met), and three PCGs (ND2, ATP8 and ND3) were initiated by ATT (Ile), respectively. However, the COI gene generally uses noncanonical initial codons across different insect groups. The use of non-canonical initial codons for the COI gene has been reported in a number of other insect species. For example, Junqueira et al (2004) and Friedrich & Muqim (2003) proposed AAA or TCG as the initial site for COI in dipteranChrysomya chloropygaand in coleopteranTribolium castanaeum, respectively. Other studies have determined that TTG is the initiation codon for COI in some invertebrates such asAnopheles quadrimaculatus(Mitchell et al, 1993),Pyrocoelia rufa(Bae et al, 2004),Caligula boisdnvalii(Hong et al, 2008) andAcraea issoria(Hu et al, 2010). In addition, the tetranucleotide TTAG inCoreana raphaelis(Kim et al, 2006), the hexanucleotide TATTAG inOstrinia nubilalisandOstrinia furnicalis(Coates et al, 2005), TTTTAG inBombyx mori(Yukuhiro et al, 2002), ATTACG inPapilio xuthus(Feng et al, 2010), and TTAAAG inPieris rapae(Mao et al, 2010) have also been proposed as the COI start codon. In the case ofA. hyperbius, we tentatively presumed CGA as the start codon for COI, which was congruent withParnassius bremeri(Kim et al, 2009),Eumenis autonoe(Kim et al, 2010), andHyphantria cunea(Liao et al, 2010). Besides ATN, GTN has also been reported in Heterocera as the initiation codon for some PCGs. For instance, GTG has been reported as the start codon for COII inCaligula boisduvalii(Hong et al, 2008) andEriogyna pyretorum(Jiang et al, 2009), and for ND1 inOchrogaster lunifer(Salvato et al, 2008).Furthermore, ND4 and ND4L inOchrogaster luniferuse GTT as their initiation codon.
Eleven of the 13 protein-coding genes had the common stop codon (TAA), while COI and COII terminated with a single T residue in theA. hyperbiusmitogenome. Similar cases have been found in most insect mitogenomes including all known lepidopteran mitogenomes. For example, a single T residue has been deemed the stop codon for COI, COII, ND5 and Cyt b, and a dinucleotide residue TA has been deemed the stop codon for ATP6, ND4, ND4L, ND6 inCoreana raphaelis(Kim et al, 2006); similarly, a single T has been considered the stop codon for COI, COII and ND4, while TA residue is considered the stop codon for ATP6 inHyphantria cunea(Liao et al, 2010). Incomplete stop codons produce functional stop codons in polycistronic transcription cleavage and polyadenylation processes (Ojala et al, 1981).
Three of the 13 PCGs (ATP8, ATP6, ND6) inA. hyperbiuswere flanked by other PCGs at the 3' end: ATP8-ATP6, ATP6-COIII, and ND6-Cyt b were overlapped by seven (ATGATAA), one (A) and one (A) nucleotide, respectively. The 3' end region of these three genes had the potential to form hairpin-like structures, which are crucial for precise mRNA cleavage to generate mature PCGs (Kim et al, 2006; Fenn et al, 2007).
Those genes encoded by the N strand are underlined. The tRNA genes are designated by single letter amino acid codes. L* and S* denote the tRNALeu(UUR)and tRNASer(UCN), respectively.
3.3 Transfer RNA and ribosomal RNA genes
All the tRNA genes showed typical clover structure, with the exception of the tRNASer(AGN)gene which lacks the dihydrouridine (DHU) stem and was replaced by a simple loop. This phenomenon has also been detected in other insect groups (Wolstenholme, 1992) including lepidopterans (Hong et al, 2008; Kim et al, 2006; Salvato et al, 2008; Liao et al, 2010). Seventeen tRNA genes had a total of 26 pair mismatches in their stems, among which eighteen G·U, seven U·U, and one A·C were present. These mismatches found in tRNAs can be corrected through RNA-editing mechanisms (Lavrov et al, 2000). To date, however, these modifications in insect tRNA genes are not well understood in light of their mechanism, although some researchers propose there to be a connection with rapid species evolution of insects (Takashi et al, 1991; Watanabe & Watanabe , 1994).
Two rRNA genes were in the observed size range of known lepidopteran mitogenomes. For example, the 1 330 bp lrRNA was well within the range of other known lepidopterans (from 1 319 bp inA. melete(Hong et al, 2009) to 1 426 bp inH. cunea(Liao et al, 2010)). The case was similar with srRNA, in which size was also within the observed size range of other lepidopteran insects (from 434 bp inOstrinia nubilalis(Coates et al, 2005) to 808 bp inH. cunea)).
3.4 Intergenic spacer sequences
Because of their rapid evolutionary rates, intergenic spacer sequences (IGS) show remarkable differences even among closely related insect species. Except for the A+T-rich region, theA. hyperbiusmitogenome in this study was interleaved with 11 intergenic spacers totaling 96 bp and ranging in size from 2−52 bp (Fig. 1). The longest spacer (52 bp) located between the tRNAGlnand ND2 genes is a common feature to all lepidopteran mitogenomes, but has not yet been detected in nonlepidopteran species. This spacer showed a relatively high level of homology (62%) with its ND2 gene, which is similar to the 70% detected inParnassius bremeri(Kim et al, 2009) but significantly different from the 32% inSasakia charonda(unpublished, NC_014224). Accordingly, this spacer is thought to have originated from a partial duplication of the ND2 gene and undergone rapid sequence divergence for their noncoding nature among even closely related taxa (Kim et al, 2009). The other IGS more than 10 bp was present between the ND5 and tRNAHis, and this 15 bp long intergenic spacer exists in 15 of the 27 determined lepidopteran mitogenomes. Furthermore, a relatively conservative element of the nucleotides ATTTT was present within this spacer, which has also been found in determined insect species in the overwhelming majority of conditions. The IGS between tRNASer(UCN) and ND1 is common among lepidopteran insects, spanning from 9 bp inDiatraea saccharalis(unpublished, NC_013274) to 38 bp inOstrinia nubilalis(Coates et al, 2005). In the present study, however, it wsa nearly absent inA. hyperbiuswith only a 2 bp overlap, which is similar to findings onAcraea issoria(Hu et al, 2010),Sasakia charonda(unpublished, NC_014224), andCalinaga davidis(Xia et al, HQ658143) with 2-, 1-, 1- overlaps respectively. The conserved ATACTAA motif is regarded as a possible recognition site for the transcription termination peptide (mtTERM protein) and is usually located in the IGS between the tRNASer(UCN) and ND1 genes. However, this motif was detected within the NDI genes ofA. hyperbius. This is same asS. charondaandC. davidis, but it is present within the tRNASer(UCN) inEumenis autonoe(Kim et al, 2010) and absent in theSasakia charonda kuriymaensis(unpublished, NC_014223).
3.5 A+T-rich region
The A+T-rich region harbors the origin sites for transcription and replication (Taanman, 1999). InDrosophilaspecies, this region includes the replication origin for mtDNA heavy-strands and minor-strands (Clary & Wolstenholme, 1987). Saito et al (2005) precisely determined that the replication origin site for mtDNA minor-strand was located in this region inBombyx mori(Yukuhiro et al, 2002). In the present study, the A+T-rich region of theA. hyperbiusmitochondrial genome was located between the srRNA and tRNAMetgenes (Tab. 2) and was 349 bp in length. This was well within the range observed in the completely sequenced lepidopteran insects from 317 bp inMelanitis leda(unpublished, by our lab) to 747 bp inBombyx mandarina(Liao et al, 2010). The A+T-rich region exhibited a remarkably high A+T content (95.41%) and did not contain macrorepeat units. However, it included some microsatellite-like repeats (e.g. polyT, (AT)9, (TA)8and poly-A), as seen in other insect species. For example, the polyT stretch (20 bp), which is considered the structural signal for recognizing proteins in the mtDNA minor-strand initiation (Kim et al, 2009), was located 24 bp downstream from srRNA preceded by the motif ATAGA, which is conserved across the lepidoptera orders as well. The microsatellite-like repeat (AT)9element, located 235 bp downstream from srRNA, was preceded by the conserved motif ATTTA, which is similar to ATTTA(TA)8inManduca sexta(Cameron et al, 2008), ATTTA(AT)8inHyphantria cunea(Liao et al, 2010), ATTTA(AT)7inCoreana raphaelis(Kim et al, 2006), and ATTTA(AT)9inPieris rapae(Mao et al, 2010). Thus, this phenomenon may be characteristic of the insect AT-rich regions. Additionally, another microrepeat unit (TA)8and a 5 bp long poly-A stretch were situated at the 284 bp site downstream from srRNA, and immediately upstream tRNAMet, respectively.
3.6 Sequence variation and codon usage
The AT-skewness values of the J strand (majority or heavy strand) and N strand (minority or light strand) were −0.135 and −0.163, respectively, indicating the occurrence of more Ts than As in both the J and N strands; whereas, the GC skewness about the J and N strands were −0.149 and 0.322, respectively, suggesting a contrary condition of Gs and Cs.
For the 13 PCGs, the A+T content at the third codon position (92.9%) was higher than the first (74.5%) and second position (70.8%). The value of the A+T content of PCGs was 79.4% with a strong A+T bias. This result has been observed in other insects species, for examples, the AT contents ofSasakia charonda,Coreana raphaelis,Parnassius bremeriandHelicoverpa armigeraPCGs have been reported to be 78.2%, 81.5%, 80.1% and 79.4%, respectively.
The relative synonymous codon usage (RSCU) analysis showed that TTA, ATT, TTT, and ATA were the four most frequently used codons, accounting for 40.4% of all codons in theA. hyperbiusmitogenome. These four codons were all composed of A or T nucleotides, which indicated their biased usage. Such results have also been detected in other sequenced lepidopteran insects. For example, these four codons account for 39.1% inTeinopalpus aureus, 44.1% inCoreana raphaelis, and 40.7% inHelicoverpa armigera. For amino acids, the Leu, Ile, Phe, and Ser were the most frequently used in theA. hyperbiusmitogenome PCGs, which is in agreement with findings for other lepidopteran insects (Fig. 3). The total number of nonstop codons (CDs) for theA. hyperbiusmitochondrial PCGs was 3 718, which accords with the range for other known butterfly species, from 3 695 inSasakia charondato 3 737 inCalinaga davidis. The codons per thousands codons(CDspT) of the Ile, Leu2 and Phe were more than 100, the CDspT of Met, Asn (asparagine), Gly (glycine), Ser2 and Tyr (tyrosine) were more than 50, and the Arg (arginine), Asp (aspartic acid), Glu (glutamic acid), Gln (glutamine), His (histidine) and Leu1 were below 20, with Cys (cysteine) the lowest at 8.61 inA. hyperbiusmitochondrial PCGs. Both the CDs and CDspT of theA. hyperbiusin this study shared similar patterns with those of other Papilionoidea butterfly species (Fig. 3).
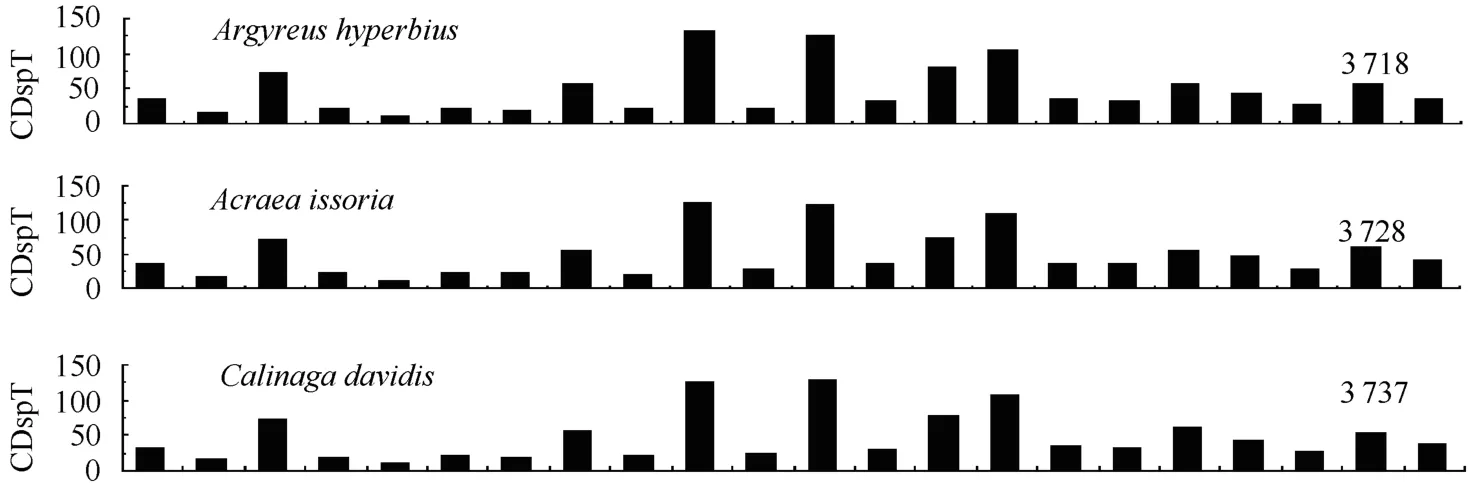
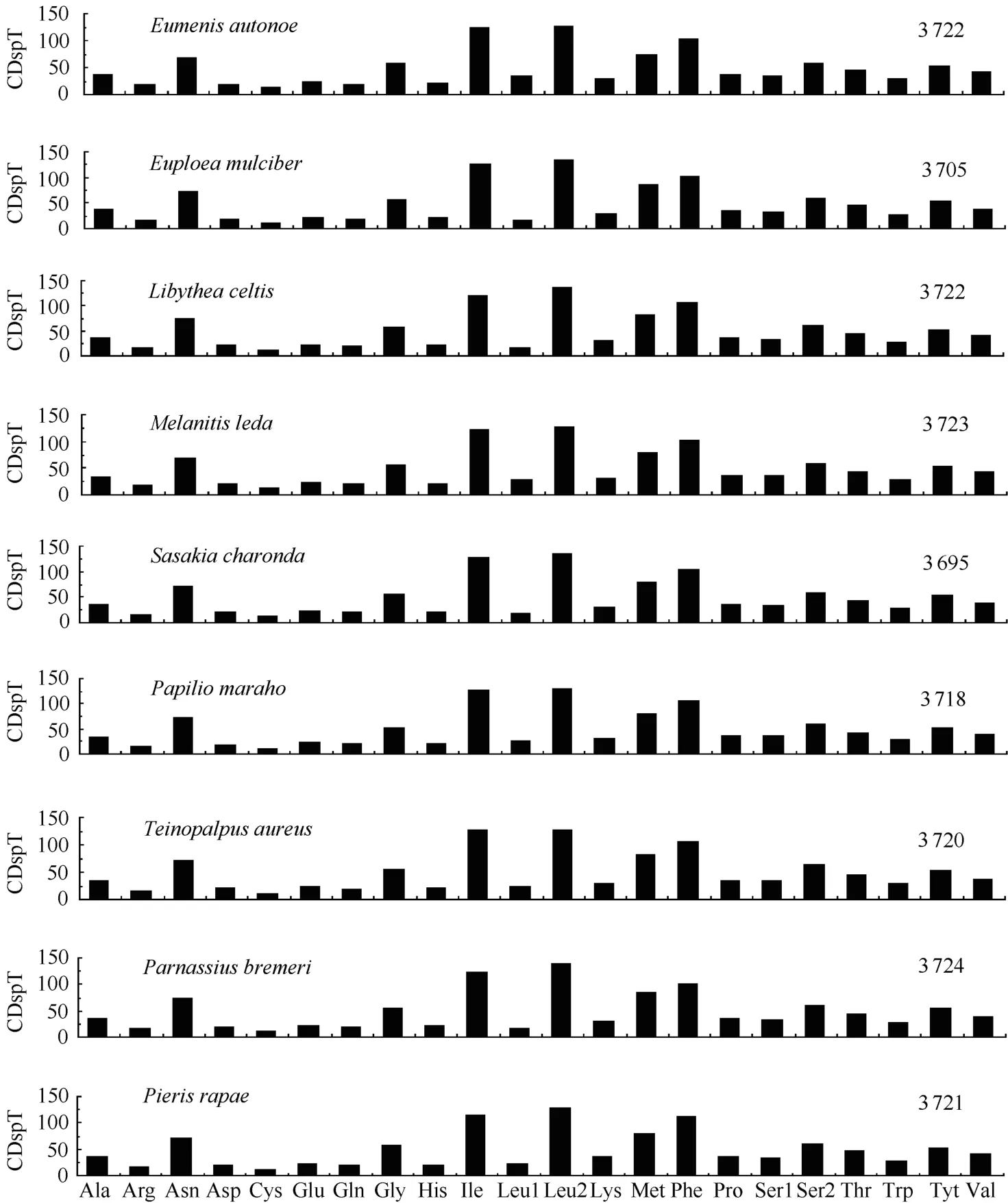
Fig. 3 Codon distribution in Papilionoidea mtDNAs
Boore JL, Lavrov D, Brown WM. 1998. Gene translocation links insects and crustaceans [J].Nature, 393: 667-668.
Bae JS, Kim I, Sohn HD, Jin BR. 2004. The mitochondrial genome of the firefly,Pyrocoelia rufa: complete DNA sequence, genome organization, and phylogenetic analysis with other insects [J].Mol Phylogenet Evol, 32: 978-985
Cameron SL, Whiting MF. 2008. The complete mitochondrial genome of the tobacco hornworm,Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths [J].Gene, 408: 112-123.
Clary DO, Wolstenholme DR. 1987.Drosophilamitochondrial DNA: conserved sequences in the AT-rich region and supporting evidence for a secondary structure model of the small ribosomal RNA [J].J Mol Evol,25: 116-125.
Coates BS, Sumerford DV, Hellmich RL, Lewis LC. 2005. Partial mitochondrial genome sequences ofOstrinia nubilalisandOstrinia furnicalis[J].Int J Biol Sci, 1: 13-18.
DeVries PJ. 2001. Nymphalidae. In: Levin SA (ed). Encyclopedia of Biodiversity[M]. Academic Press.
Fenn JD, Cameron SL, Whiting MF. 2007. The complete mitochondrial genome of the Mormon cricket (Anabrussimplex: Tettigoniidae: Orthoptera) and an analysis of control region variability [J].Insect Mol Biol, 16: 239-252.
Feng X, Liu DF, Wang NX, Zhu CD, Jiang GF. 2010. The mitochondrial genome of the butterflyPapilio xuthus(Lepidoptera: Papilionidae) and related phylogenetic analyses [J].Mol Biol Rep, 37: 3877-3888.
Friedrich M, Muqim N. 2003. Sequence and phylogenetic analysis of the complete mitochondrial genome of the flour beetleTribolium castanaeum[J].Mol Phylogenet Evol, 26(3):502-512.
Hao JS, Su CY, Zhu GP, Chen N, Wu DX, Zhang XP. 2007. The molecular morphologies of mitochondrial 16S rDNA of the main butterfly lineages and their phylogenetic significances [J].Genet Mol Biol, 18(2): 111-123.
Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT [J].Nucleic Acid Symp Ser, 41: 95-98.
Hong G, Jiang S, Yu M, Yang Y, Li F, Xue F, Wei Z. 2009. The complete nucleotide sequence of the mitochondrial genome of the cabbage butterfly,Artogeia melete(Lepidoptera: Pieridae) [J].Acta Biochim Biophys Sin,41: 446-455.
Hong MY, Lee EM, Jo YH, Park HC, Kim SR, Huang JS, Jin BR, Kang PD, Kim K, Han YS, Kim I. 2008. Complete nucleotide sequence and organization of the mitogenome of the silk mothCaligula biosducalii(Lepidoptera: Saturniidae) and comparison with other lepidopteran insects [J].Gene, 413: 49-57.
Hu J, Zhang DX, Hao JS, Huang DY, Cameron S, Zhu CD. 2010. The complete mitochondrial genome of the yellow coaster,Acraea issoria(Lepidoptera: Nymphalidae: Heliconiinae: Acraeini): sequence, gene organization and a unique tRNA translocation event [J].Mol Biol Rep, 37(7): 3431-3438.
Irwin DM, Kocher TD, Wilson AC. 1991. Evolution of the cytochrome b gene of mammals [J].Mol Evol, 32:128-144.
Jiang ST, Hong GY, Yu M, Li N, Yang Y, Liu YQ, Wei ZJ. 2009. Characterization of the complete mitochondrial genome of the giant silkworm moth,Eriogyna pyretorum(Lepidoptera: Saturniidae) [J].Int J Biol Sci, 5(4): 351-365.
Junqueira ACM, Lessingera AC, Torresa TT, Silvab FR, Vettorec AL, Arrudad P, Espin AMA. 2004. The mitochondrial genome of the blowflyChrysomya chloropyga(Diptera: Calliphoridae) [J].Gene, 339: 7-15.
Kim I, Lee EM, Seol KY, Yun EY, Lee YB, Hwang JS, Jin BR. 2006. The mitochondrial genome of the Korean hairstreak,Coreana raphaelis(Lepidoptera: Lycaenidae) [J].Insect Mol Biol, 15: 217-225.
Kim MI, Beak JY, Kim MJ, Jeong HC, Kim KJ, Bae CH, Han YS, Jin BR, Kim I. 2009. Complete nucleotide sequence and organization of the mitogenome of the red-spotted Apollo butterfly,Parnassius bremeri(Lepidoptera: Papilionidae) and comparison with other lepidopteran insects [J].Mol Cell, 28: 347-363.
Kim MJ, Wan XL, Kim KG, H JS, Kim I. 2010. Complete nucleotide sequence and organization of the mitogenome of endangeredEumenis autonoe(Lepidoptera: Nymphalidae) [J].Afr J Biotechnol, 9 (5): 735-754.
Kocher TD, Thomas WK, Meyer A, Edwards SV, Paabo S, Villablanca FX, Wilson AC. 1989. Dynamics of mitochondrial DNA evolution in animals: Amplification and sequencing with conserved primers [J].Proc Natl Acad Sci USA, 86: 6196-6200.
Kumar S, Tamura K, Nei M. 2004. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment [J].Brief Bioinform, 5: 150-163.
Lavrov DV, Brown WM, Boore JL. 2000. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipedeLithobius forficatus[J].Proc Natl Acad Sci USA, 97: 13738-13742.
Liao F, Wang L, Wu S, Li YP, Zhao L, Huang GM, Niu CJ, Liu YQ, Li MG. 2010. The complete mitochondrial genome of the fall webworm,Hyphantria cunea(Lepidoptera: Arctiidae) [J].Int J Biol Sci, 6(2):172-186.
Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence [J].Nucleic Acids Res, 25: 955-964.
Mao ZH, Hao JS, Zhu GP, Hu J, Si MM, Zhu CD. 2010. Sequencing and analysis of the complete mitochondrial genome ofPeris rapaeLinnaeus (Lepidoptera: Peridae) [J].Acta Entomol Sin, 53(11): 1295-1304.
Mitchell SE, Cockburn AF, Seawright JA. 1993. The mitochondrial genome ofAnopheles quadrimaculatusspecies A: complete nucleotide sequence and gene organization [J].Genome, 36: 1058-1073.
Ojala D, Montoya J, Attardi G. 1981. tRNA punctuation model of RNA processing in human mitochondria [J].Nature, 290: 470-474.
Saito S, Tamura K, Aotsuka T. 2005. Replication origin of mitochondrial DNA in insects [J].Genetics, 171(4): 1695-1705.
Salvato P, Simonato M, Battisti A, Negrisolo E. 2008. The complete mitochondrial genome of the bag-shelter mothOchrogaster lunifer(Lepidoptera, Notodontidae) [J].BMC Evol Biol, 9:331.
Simon C, Frati F, Bekenbach A, Crespi B, Liu H, Flook P. 1994. Evolution, weighting, and phylogenetic utility of mitochondrial genesequences and a compilation of conserved polymerase chainreaction primers [J].Ann Entomol Soc Am, 87: 651-701.
Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT. 2006. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA [J].Ann Rev Ecol Evol Syst, 37: 545-579.
Simons RB, Weller SJ. 2001. Utility and evolution of cytochrome b in insects [J].Mol Phylogenet Evol, 20: 196-210.
Simonsen TJ, Wahlberg N, Brower AVZ, Jong R. 2006. Morphology, molecules and fritilllaries: approaching a stable phylogeny for Argynnini (Lepidoptera: Nymphalidae)[J].Insect Syst Evol, 37: 405-418.
Singh VK, Mangalam AK, Dwivedi S, Naik S. 1998. Primer premier: Program for design of degenerate primers from a protein sequence [J].Biol Techniques, 24:318-319.
Taanman JW. 1999. The mitochondrial genome: structure, transcription and replication [J].Biochem Biophys Acta, 1410: 103-123
Takashi Y, Yohichi W, Kimitsuna W. 1991. A novel clover leaf structure found in mammalian mitochondrial tRNASer[J].Nucl Acid Res, 19: 6101-6105.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The Clustal X windows interface: flexible strategies for multiple sequences alignment aided by quality analysis tools [J].Nucl Acids Res, 24: 4876-4882.
Watanabe Y, Watanabe K. 1994. Higher order structure of bovine mitochondrial tRNASerUGAchemical modification and computer modeling [J].Nucleic Acids Res, 22: 5378-5384.
Wolstenholme DR. 1992. Animal mitochondrial DNA: structure and evolution [J].Int Rev Cytol, 141:173-216.
Wu Q. 2008.Argyreus hyperbius[J].Chn Nat, 1: 74-77.(in Chinese)
Xia J, Hu J, Zhu GP, Zhu CD, Hao JS. 2011. Sequencing and analysis of the complete mitochondrial genome ofCalinaga davidisOberthür (Lepidoptera: Nymphalidae) [J].Acta Entomol Sin, 54(5): 555-565.
Yamauchi MM, Miya MU, Nishida M. 2004. Use of a PCR-based approach for sequencing whole mitochondrial genomes of insects: two examples (cockroach and dragonfly) based on the method developed for decapod crustaceans [J].Insect Mol Biol, 13: 435-442.
Yukuhiro K, Sezutsu H, Itoh M, Shimizu K, Banno Y. 2002. Significant levels of sequence divergence and gene rearrangements have occurred between the mitochondrial genomes of the wild mulberry silkmoth,Bombyx mandarinaand its close relative, the domesticated silkmoth,Bombyx mori[J].Mol Biol Evol, 19: 1385-1389.
斐豹蛱蝶线粒体基因组全序列的测定和分析
王晓灿1, 孙晓燕2, 孙倩倩1, 张大秀1, 胡 静1, 杨 群2,*, 郝家胜1,2,*
(1.安徽师范大学 生命科学学院分子进化与生物多样性研究室,安徽 芜湖241000;
2.中国科学院南京地质古生物研究所 现代古生物学与地层学国家重点实验室,江苏 南京210008)
该研究对斐豹蛱蝶(Argyreus hyperbius)(鳞翅目:蛱蝶科)线粒体基因组全序列进行了测定和初步分析。结果表明:斐豹蛱蝶线粒体基因全序列全长为15 156bp, 包含13个蛋白质编码基因、22个tRNA和2个rRNA基因以及1个非编码的A+T富集区, 基因排列顺序与其它鳞翅目种类一致; 线粒体全序列核苷酸组成和密码子使用显示出明显的A+T偏好(80.8%)和轻微的AT 偏移(AT skew, −0.019)。基因组中共存在11个2~52 bp不等的基因间隔区, 总长96 bp; 以及14个1~8 bp不等的基因重叠区, 总长34 bp。除COI以CGA作为起始密码子外, 13个蛋白质编码基因中的其余12个基因是以ATN作为起始密码子。除COI和COII基因是以单独的一个T为终止密码子, 其余11个蛋白质编码基因都是以TAA结尾的。除了缺少DHU臂的tRNASer(AGN), 其余的tRNA基因都显示典型的三叶草结构。tRNA(AGN)和ND1之间的基因间隔区包含一个ATACTAA结构域, 这个结构域在鳞翅目中是保守的。A+T富集区没有较大的多拷贝重复序列, 但是包含一些微小重复结构:ATAGA结构域下游的20 bp poly-T结构, ATTTA结构域后的(AT)9重复, 以及位于tRNAMet上游的5 bp poly-A结构等。这项研究所揭示的斐豹蛱蝶的线粒体基因组特征, 不仅为认识蛱蝶科的遗传多样性贡献数据, 而且对于该物种的保护生物学、群体遗传学、谱系地理及演化研究等具有重要意义。
2011-04-11;接受日期:2011-07-01
安徽省高校省级自然科学研究重点项目(KJ 2010A 142);中国科学院知识创新工程重要方向项目( KZCX22YW2JC104);现代古生物学和地层学国家重点实验室开放基金(104143)
斐豹蛱蝶; 蛱蝶科; 鳞翅目; 线粒体基因组
Q969.42; Q969.439.2
A
0254-5853-(2011)05-0465-11
10.3724/SP.J.1141.2011.05465
date: 2011-04-11; Accepted date: 2011-07-01
s: This work was supported by the Provincial Key Project of the Natural Science Foundation from Anhui Province, China (KJ2010A142), the Chinese Academy of Sciences (KZCX22YW2JC104), the CAS/SAFEA International Partnership Program for Creative Research Teams, and the State Key Laboratory of Palaeobiology and Stratigraphy, Nanjing Institute of Geology and Palaeontology, Chinese Academy of Sciences (104143)
*Corresponding authors (通信作者), E-mail: qunyang@nigpas.ac.cn; jshaonigpas@sina.com
