5p部分单体3例全基因组拷贝数变异分析
2011-01-19倪锦文詹国栋王慧君黄国英周文浩
杨 琳 倪锦文 詹国栋 王慧君 陈 超 黄国英 周文浩
Cri du chat综合征(猫叫综合征、5p缺失综合征)在活产婴儿中发病率为1/15 000~1/50 000,见于所有种族的人群[1],其中约1%患儿有严重智力低下(IQ<20)[1]。Cri du chat综合征是由于5号染色体短臂末端的缺失所致,但缺失的片段大小在不同患儿中相差很大,最小可仅包含5p15.2,最大可累及整个短臂。有研究认为p15.2缺失与发育迟缓相关,p15.3缺失与猫叫样哭声和语言发育障碍相关,且大片段的缺失引起的智力低下和发育迟缓更为严重[2]。但也有学者提出除片段大小外,还有其他因素决定表型[3],缺失片段位于末端(5p15.33),包含端粒逆转录酶基因(TERT),可能与表型相关[4]。也有研究提示位于p15.2的CTNND2基因的缺失与严重智力障碍有关[5]。但由于之前研究技术所限,定位欠精确,并且在缺失范围内其他基因的作用、在缺失范围外其他拷贝数变异(CNVs)的作用尚不清楚。
2004年CNVs报道以来[6],因其在基因组中覆盖的核苷酸总数远大于单核苷酸多态性(SNP)而倍受关注,CNVs正不断被用以解释很多复杂疾病,如智力低下、孤独症和多发畸形等。比较基因组杂交芯片(CGH)和SNP芯片技术的应用日趋增多,使得疾病关键区域的定位更为准确,建立精确表型基因型关联性的可能性不断增加。本研究的目的在于通过对具有部分相同基因型(5p部分缺失)病例的分析,建立罕见潜在致病性CNVs合理的筛选和分析流程,进一步解释Cri du chat综合征的表型基因型关联性。
1 方法
1.1 病例来源 2009年6月至2010年5月复旦大学附属儿科医院新生儿病房收治的3例染色体核型分析提示为Cri du chat综合征的患儿。
1.2 伦理学 本研究经复旦大学附属儿科医院伦理委员会批准,患儿家属签署知情同意书后进入本研究。
1.3 临床资料采集 包括:一般情况(性别、胎龄和出生体重等)、Cri du chat综合征特征性表现(哭声等)、特殊面容、指趾畸形、合并先天性疾病的情况等。
1.4 全基因组CNVs检测
1.4.1 DNA提取 取外周静脉血经EDTA抗凝,使用FUJI公司QuickGene-Mini80及DNA全血试剂盒,从全血样本中提取DNA。使用NanoDrop ND-1000分光光度计检测DNA浓度及吸光度值,电泳检测DNA降解程度。
1.4.2 芯片检测 取基因组DNA 3~5 μg进行微阵列芯片检测。采用Affymetrix公司Cytogenetic Whole-Genome 2.7M 芯片。采用Chromosome Analysis Suite (ChAS)软件进行数据结果的分析。
1.5 CNVs片段的筛选步骤 ①ChAs软件分析的结果中,选择重复片段>150 kb,缺失片断>50 kb的CNVs数据进行下一步分析;②对照原始图像除外假阳性结果;③除外在国际基因组拷贝数变异多态性数据库(DGV)中报道为正常人群的CNVs(片段至少有80%以上的重合,且重复或缺失类型相同);④除外片段区域内不包含基因的CNVs。
1.6 临床表型基因型关联性分析 搜索DECIPHER数据库报道的5p部分缺失病例(不包含除5p外的CNVs),CNVs片段有较大重合病例的临床表现及基因型信息,结合本研究3例与DECIPHER数据库已报道的Cri du chat综合征患儿的临床表型,进行5p缺失大小及范围分析,对重复区域行候选基因分析。
2 结果
2.1 CNVs筛选 分析ChAs软件结果中,选取重复片段>150 kb,缺失片段>50 kb的CNVs,共29个CNVs片段,除外4个假阳性CNVs、11个DGV上报道为正常人群CNVs和3个不包含基因的CNVs,获得罕见具有潜在临床意义的CNVs共11个(图1)。
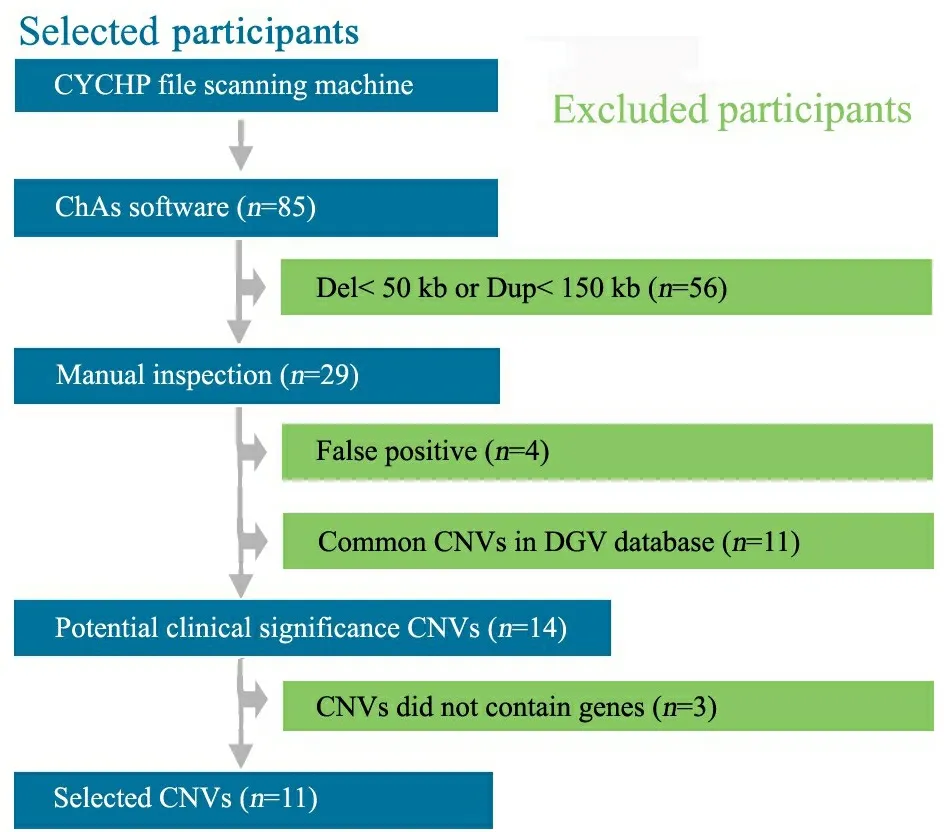
图1 CNVs的筛选步骤
Fig 1 Flow chart of CNVs selection procedure
11个CNVs片段大小在66~31 328 kb,其中缺失为9个(81.2%),重复为2个(18.2%)。例1有3个CNVs位于5p,且位置相近,故连接为1个片段进行分析;例2有4个CNVs位于5p,且位置相近,连接为1个片段;1个CNVs位于9p;例3有2个CNVs位于5p,且位置相近,连接为1个片段;1个CNVs位于7p。最终得到5个CNVs,基本特征见表1,缺失或重复情况见图2。
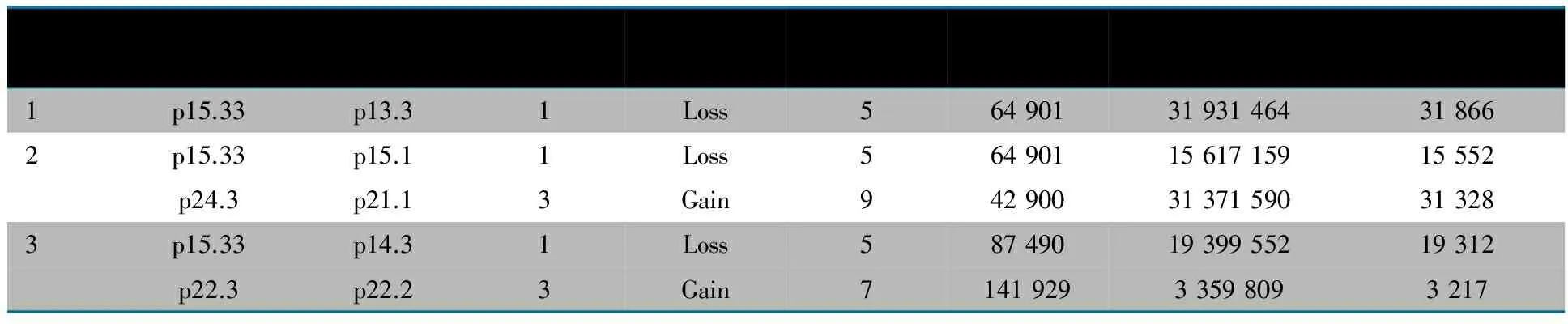
表1 3例Cri du chat综合征患儿筛选出的CNVs的基本特征
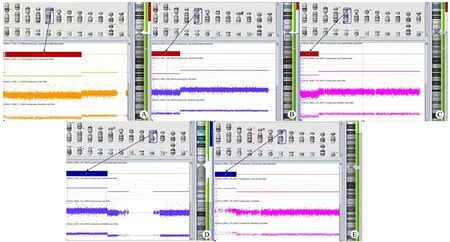
图2 3例Cri du chat综合征患儿的缺失和重复
Fig 2 Deletions and duplications of 3 patients by Affymetrix array
Notes Each probe represented a single dot and plots on X axis according to its genome position.Duplication/deletion was shown respectively as up/down dot according to the base line.The blue/red bar represented the affected region.A: 31 866 kb deletion in 5p15.33-13.3 in case 1; B: a 15 552 kb deletion in 5p15.33-15.1 in case 2; C: 31 328 kb duplication in 9p24.3-21.1 in case 2; D: 19 312 kb deletion in 5p15.33-14.3 in case 3; E: 3 217 kb duplication in 7p22.3-22.2 in case 3
2.2 临床表型 例1为女性,例2和3均为男性。例1有宫内生长发育迟缓、Cri du chat综合征特征性的高调哭声、先天性心脏病(右室双出口、室间隔缺损、动脉导管未闭和肺动脉高压)和指趾畸形;例2有宫内生长发育迟缓、先天性喉软骨发育不良、特殊面容(眼距过宽、双眼下斜和耳位异常)、末指两节畸形和单一掌纹畸形(通贯掌)(图3);例3有哭声异常(为外院转入病例,描述为生后哭声异常)、高腭弓,有先天性巨结肠(表2)。
DECIPHER数据库检索到CNVs片段与本组病例较大重合的Cri du chat综合征患儿有5例(编号为249537、249606、249117、249796和251341),其临床表型如表1所示,其中4例存在发育迟缓或智力低下,1例有Cri du chat综合征特征性的高调哭声,3例(编号为249796、249537和249606)表现为鼻音重,2例(编号为249117和249537)有特殊面容,表现为眼睑过小、眼睛深陷和小耳,伴指趾畸形。
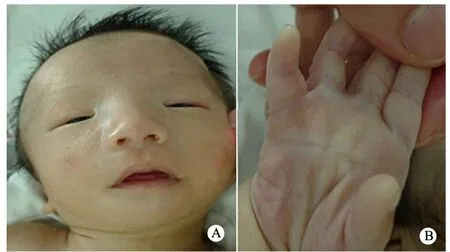
图3 例2患儿特殊面容及掌指畸形
Fig 3 The facial features and deformities of palma and finger in case 2
Notes A:facial features of case 2 including hypertelorism, blepharo-phimosis, low set right ear; B: two sections of right little finger, single palmar crease
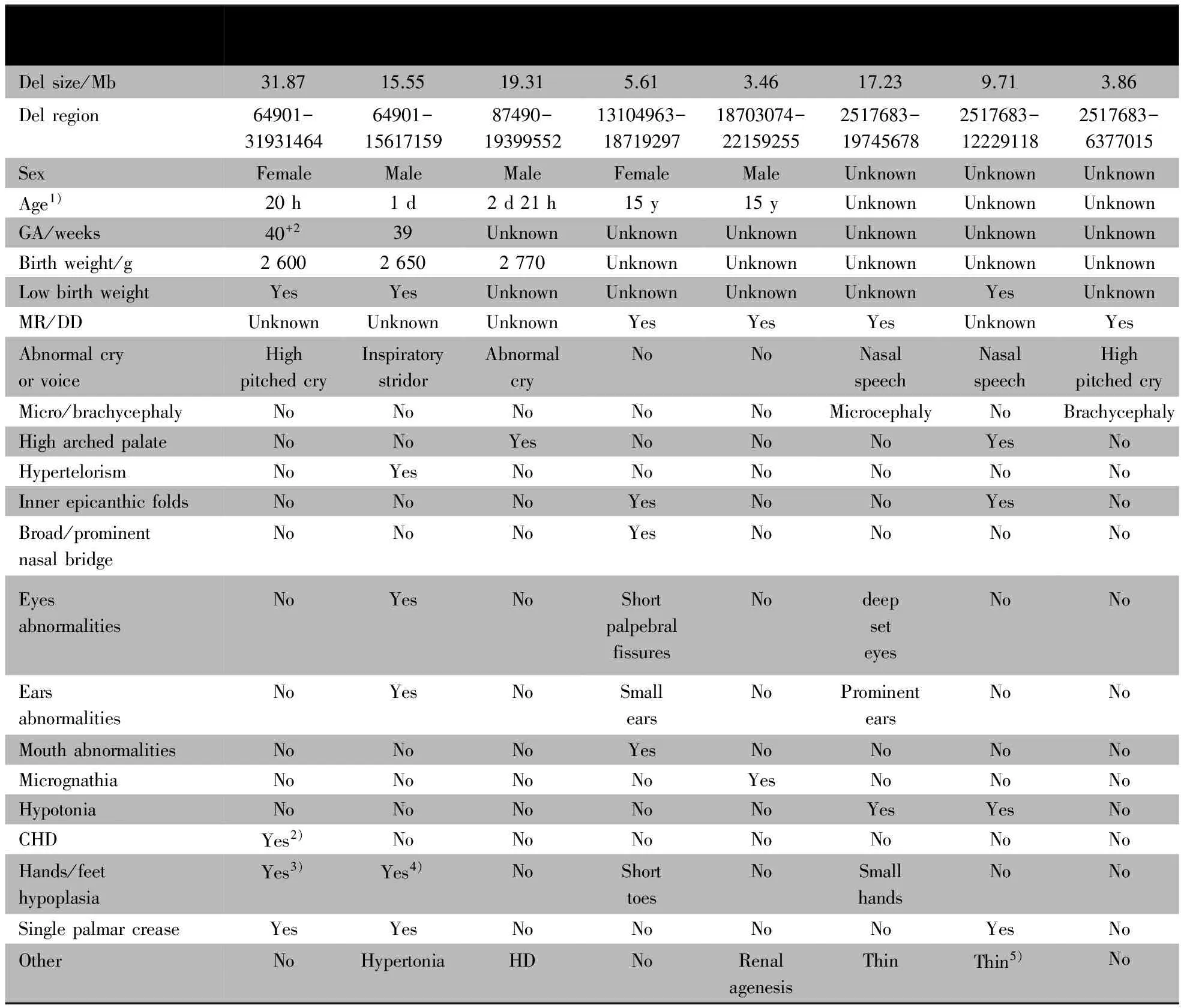
表2 本研究3例和DECIPHER数据库5例Cri du chat综合征的临床表现
Notes GA:gestational age; MR/DD:mental retardation/developmental delay; CHD: congenital heart disease; HD:hirschsprung's disease; 1)at initial presentation; 2)two outflows of right ventical; 3)three sections of right thumb;4)two sections of little finger;5) thin upper lip,thin/long face
2.4 临床表型基因型相关性分析 本组3例和DECIPHER数据库中5例Cri du chat综合征患儿的5p缺失大小及位置见图4,区域1为异常哭声的重复区域,区域2为异常面容的重复区域。进一步分析重复区域1的基因,覆盖IRX1、IRX2和ADAMTS16等基因(图5),重复区域2覆盖DNAH5、TRIO和ANKH等基因(图6)。
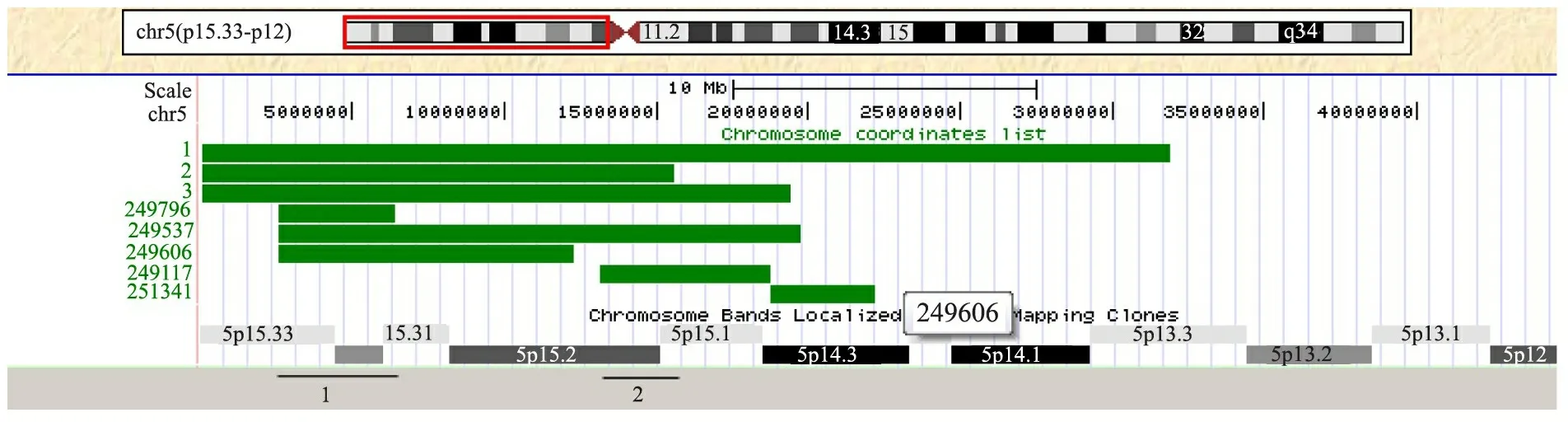
图4 本组3例及DECIPHER数据库中5例Cri du chat综合征患儿5p缺失大小及范围
Fig 4 Characteristics of the 5p deletion and sizes of both 3 cases and 5 DECIPHER patients
Notes 1: overlapping region of cases with abnormal cry or voice; 2: overlapping region of cases with facial deformation

图5 重合区域1范围内包含的候选基因
Fig 5 Covered candidate genes in the overlapping region 1
Notes:In the top panel, an ideogram showed deletion band of chromosome 5p15.33-15.31.In the middle panel, six green bars represented overlapping deleted region in six cases.Bottom panel showed affected genes in this region
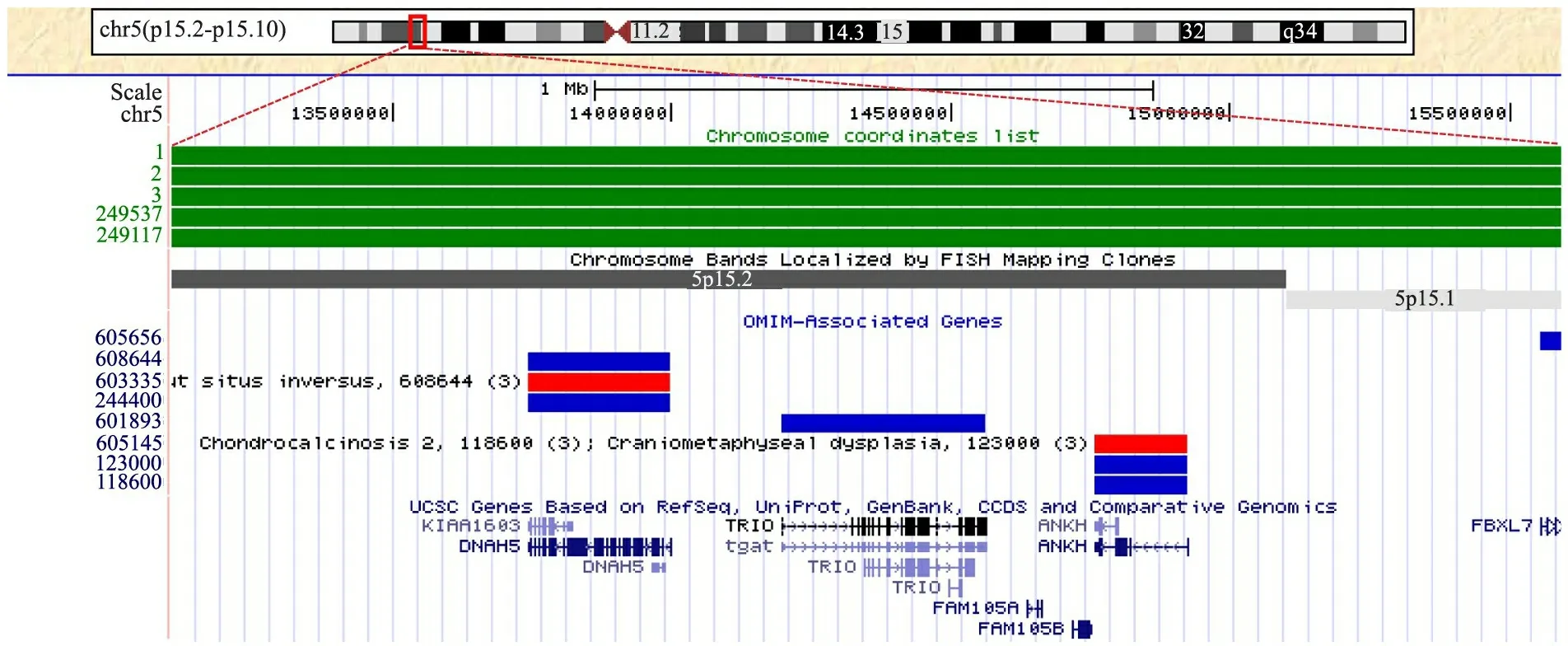
图6 重合区域2范围内包含的候选基因
Fig 6 Covered candidate genes in the overlapping region 2
Notes In the top panel, an ideogram showed deletion band of chromosome 5p15.2-15.1.In the middle panel, five green bars represented overlapping deleted region in five cases.Bottom panel showed affected genes in this region.The blue bars represented OMIM genes and red bars represented morbid OMIM genes
3 讨论
Cri du chat综合征的典型临床表现有:低出生体重和生长迟缓、智力发育迟缓和运动发育延迟;哭声高调、尖锐似猫叫;小头、小下颌畸形、眼距宽、内眦皱褶、眼睛下斜、耳低位或形状异常;并指(趾)和异常掌纹(通贯掌)[9]。15%~20%患儿伴有先天性心脏病[10]。在新生儿,典型猫叫样哭声是最具特征性的表现,通常在数个月或数年后消失。也有不具备此特征性表现患儿的报道,且猫叫样哭声还可见于其他神经系统疾病,故而不能作为特征性诊断指标。此外缺氧发作、青紫和吸气性喉鸣等表现亦见于部分Cri du chat综合征病例。本病多数发生于卵子或精子的发育过程或胚胎发育早期,仅10%的患儿是由于父母一方携带有5号染色体的平衡易位[9]。
本组3例均表现为哭声异常,表现为高调哭声或哭声弱伴吸气性喉鸣,DECIPHER数据库中5例患儿有3例表现为高调哭声或鼻音重。Zhang等[18]报道特殊哭声的关键区域位于5p15.31的末端;Church等[19]报道症状不典型的5p部分单体患儿猫叫样哭声关键区域位于5p15.2末端;Mainardi等[3]报道典型猫叫样哭声的关键区域位于5p15.3。本研究基因分析结果显示,重合区域为5p15.33-15.31,大小为3.86 Mb,与上述文献报道并不一致。5p15.33-15.31可能为哭声和声音异常的关键区域,覆盖基因IRX1、IRX2和ADAMTS16等候选基因。IRX1和IRX2均属于Iroquois同源基因家族,该家族的基因与脊椎动物的胚胎形成相关。动物模型研究发现,该基因家族与胚胎神经发育过程中的前-后轴、背-腹轴模式化的形成相关,并与耳的发育及肢体的形成相关[11]。目前未见与咽、喉部等影响哭声及构成发音的部位发育相关的报道。ADAMTS16基因编码产物为锌依赖的金属蛋白酶,主要表达于肾脏,为血压调节的候选基因[12],未见与发育等相关报道。
本研究1例(例2)和 DECIPHER数据库2例患儿均表现为特殊面容。本研究基因分析结果显示,重叠区域为5p15.1-15.2,大小为2.51 Mb,可能为5p部分单体特殊面容的关键区域。该区域包含DNAH5、TRIO和ANKH基因。DNAH5编码微管相关动力蛋白复合物的重链部分,该基因的突变与纤毛动力缺乏相关,可引起反复呼吸道感染及精子活动力缺乏[13]。TRIO的编码蛋白质有3个功能区,1个丝氨酸/苏氨酸激酶功能区和2个鸟嘌呤交换因子功能区,这些功能区提示该蛋白可能参与多个信号通路,并调控细胞的增殖。Ferraro等[14]通过在大鼠和小鼠模型上调该基因,发现其与神经内分泌细胞中分泌型颗粒的成熟相关,这2个基因目前尚无与特殊面容相关的功能学方面的报道。ANKH编码一多通道跨膜蛋白,介导调控焦磷酸水平,故而可能参与组织钙化,并为高等动物关节炎的易感因素[15],该基因的突变与颅骨干骺端发育不良及软骨钙质沉着病相关[16,17],可能与特殊面容等表型具有相关性。此外,文献报道小头畸形和智力发育迟缓关键区域也位于5p15.2,因本研究纳入均为新生儿病例,故无法评价是否存在智力低下、生长发育迟缓和语言障碍,无法对相应的关键区域进行分析。
本研究还发现,例2除5p部分缺失外,还有9p24.3-p21.1片段的重复。9p全部或部分三体报道较少,迄今尚不足200例[20],且多数报道合并有其他染色体的缺失。9p部分或全部重复的典型表型有智力发育迟缓、小短头畸形、眼窝深、眼距宽且眼裂下斜、宽鼻、鼻尖圆、嘴角下斜、低位耳、手脚的畸形和骨龄延迟等[21~25]。有报道9p部分重复与表型相关的关键区域为9p22.1和9p22.2[26],与本研究例2缺失的区域一致。例2存在眼距宽、耳位低,与9p部分重复症状符合,但该异常面容亦为5p部分缺失的表型;例2同时有指、趾畸形,与9p重复表型相符,但不为5p部分缺失的表型。但例2尚处于新生儿期,特征性面部表现不典型,且智力、语言发育情况尚不能评估。故例2可能同时存在5p部分缺失与9p部分重复。
例3除5p部分缺失外,还发现7p22.3内有3 217 kb的重复。7p22.3为较大片段重复,检索相关文献,7p部分重复较罕见,可表现为智力发育迟缓、前囟大、眼距过宽、低位耳、腭异常、心血管异常、骨骼肌肉畸形和异常掌纹[27,,28]。本组例3的主要表型为先天性巨结肠,与既往研究报道不一致。先天性巨结肠染色体异常多见于唐氏综合征[29]、13q22(EDNRB)缺失[30]、10q11.2(RET)缺失[31]和2q22(ZFHX1B)[32]缺失相关,单基因及非综合征的先天性巨结肠与4p12(PHOX2B)、5p13.1-p12(GDNF)、20q13.2-q13.3(EDN3)、11p13(BDNF)、9p21-p12(RMRP)、22q13(SOX10)、10q11.2、19p13.3和1p36.1等多基因、多区域相关。但例3患儿的CNVs累及区域均不在相关位点中,是否这些区域亦为先天性巨结肠的关键区域或包含有关键的致病基因有待进一步研究证实。
本研究采用罕见CNVs的分析方法,得到如下结论和启示:①对于多发畸形患儿进行全基因组CNVs分析能发现常规G带染色体核型分析所不能发现的异常,尤其对于表型不典型且部分表型辨别困难的新生儿提供了早期诊断的可能;②为5p部分缺失建立表型-基因型的关联性提供帮助,并提出了新的可能与疾病相关的候选基因;③本研究报道了1例可能同时存在5p部分缺失和9p部分重复的患儿。④为先天性巨结肠提供了未报道的可能相关区域。由于病例数量较少,上述发现需要扩大样本量的验证及进一步的相关基因功能的研究。
[1]Sigafoos J, O'Reilly MF, Lancioni GE.Cri-du-chat.Dev Neurorehabil, 2009,12(3):119-121
[2]Niebuhr E.The Cri du chat syndrome: epidemiology, cytogenetics, and clinical features.Hum Genet,1978,44(3):227-275
[3]Mainardi PC, Perfumo C, Calì A, et al.Clinical and molecular characterisation of 80 patients with 5p deletion: genotype-phenotype correlation.J Med Genet, 2001,38(3):151-158
[4]Fang JS, Lee KF, Huang CT, et al.Cytogenetic and molecular characterization of a three-generation family with chromosome 5p terminal deletion.Clin Genet, 2008 ,73(6):585-590
[5]Zhang A, Zheng C, Hou M, et al.Deletion of the telomerase reverse transcriptase gene and haploinsufficiency of telomere maintenance in Cri du chat syndrome.Am J Hum Genet, 2003,72(4):940-948
[6]Medina M, Marinescu RC, Overhauser J, et al.Hemizygosity of delta-catenin (CTNND2) is associated with severe mental retardation in cri-du-chat syndrome.Genomics, 2000,63(2):157-164
[7]Iafrate AJ, Feuk L, Rivera MN, et al.Detection of large-scale variation in the human genome.Nat Genet,2004,36(9):949-951
[8]Freeman JL, Perry GH, Feuk L, et al.Copy number variation: new insights in genome diversity.Genome Res, 2006,16(8):949-961
[9]Rodríguez-Caballero A, Torres-Lagares D, Rodríguez-Pérez A, et al.Cri du chat syndrome: a critical review.Med Oral Patol Oral Cir Bucal, 2010,15(3):473-478
[10]Hills C, Moller J H, Finkelstein M, et al.Cri du chat syndrome and congenital heart disease: A review of previously reported cases and presentation of an additional 21 cases from the pediatric cardiac care consortium.Pediatrics,2006,117(5):924-927
[11]Bosse A, Zülch A, Becker MB, et al.Identification of the vertebrate Iroquois homeobox gene family with overlapping expression during early development of the nervous system.Mech Dev, 1997,69(1-2):169-181
[12]Joe B, Saad Y, Lee NH, et al.Positional identification of variants of Adamts16 linked to inherited hypertension.Hum Mol Genet,2009,18(15):2825-2838
[13]Olbrich H, Haffner K, Kispert A, et al.Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry Nat Genet,2002,30(2):143-144
[14]Ferraro F, Ma XM, Sobota JA, et al.Kalirin/Trio Rho guanine nucleotide exchange factors regulate a novel step in secretory granule maturation.Mol Biol Cell,2007,18(12):4813-4825
[15]Ho AM, Johnson MD, Kingsley DM.Role of the mouse ank gene in control of tissue calcification and arthritis.Science,2000,289(5477):265-270
[16]Nurnberg P, Thiele H, Chandler D, et al.Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia.Nat Genet,2001,28(1):37-41
[17]Pendleton A, Johnson M D, Hughes A, et al.Mutations in ANKH cause chondrocalcinosis.Am J Hum Genet,2002,71(4):933-940
[18]Zhang X, Snijders A, Segraves R, et al.High-resolution mapping of genotype-phenotype relationships in cri du chat syndrome using array comparative genomic hybridization.Am J Hum Genet, 2005,76(2):312-326
[19]Church DM, Yang J, Bocian M, et al.A high-resolution physical and transcript map of the Cri du chat region of human chromosome 5p.Genome Res,1997,7(8):787-801
[20]Zou YS, Huang XL, Ito M, et al.Further delineation of the critical region for the 9p-duplication syndrome.Am J Med Genet A, 2009,149A(2):272-276
[21]Centerwall WR, Beatty-DeSana JW.The trisomy 9p syndrome.Pediatrics,1975,56(5):748-755
[22]Wilson GN, Raj A, Baker D.The phenotypic and cytogenetic spectrum of partial trisomy 9.Am J Med Genet, 1985,20(2):277-282
[23]Smart RD, Viljoen DL, Fraser B.Partial trisomy 9-further delineation of the phenotype.Am J Med Genet, 1988,31(4):947-951
[24]Littooij AS, Hochstenbach R, Sinke RJ, et al.Two cases with partial trisomy 9p: molecular cytogenetic characterization and clinical follow-up.Am J Med Genet,2002,109(2):125-132
[25]Temtamy SA, Kamel AK, Ismail S, et al.Phenotypic and cytogenetic spectrum of 9p trisomy.Genet Couns, 2007,18(1):29-48
[26]de Ravel TJ, Fryns JP, Van Driessche J, et al.Complex chromosome re-arrangement 45,X,t(Y;9) in a girl with sex reversal and mental retardation.Am J Med Genet A, 2004,124A(3):259-262
[27]Papadopoulou E, Sifakis S, Sarri C, et al.A report of pure 7p duplication syndrome and review of the literature.Am J Med Genet A, 2006,140(24):2802-2806
[28]Cox H, Stewart H, Hall L, et al.Phenotypic spectrum of interstitial 7p duplication in mosaic and non-mosaic forms.Am J Med Genet, 2002,109(4):306-310
[29]Moore SW, Johnson AG.Hirschsprung's disease: genetic and functional associations of Down's and Waardenburg syndromes.Semin Pediatr Surg,1998,7(3):156-161
[30]Shanske A, Ferreira JC, Leonard JC, et al.Hirschsprung disease in an infant with a contiguous gene syndrome of chromosome 13.Am J Med Genet, 2001,102(3):231-236
[31]Fewtrell MS, Tam PK, Thomson AH, et al.Hirschsprung's disease associated with a deletion of chromosome 10 (q11.2q21.2): a further link with the neurocristopathies?J Med Genet, 1994,31(4):325-327
[32]Amiel J, Espinosa-Parrilla Y, Steffann J, et al.Large-scale deletions and SMADIP1 truncating mutations in syndromic Hirschsprung disease with involvement of midline structures.Am J Hum Genet, 2001,69(6):1370-1377
