儿童常染色体显性遗传Emery-Dreifuss肌营养不良症1例并文献复习
2011-01-19张礼萍吕俊兰吴沪生
伍 妘 张礼萍 吕俊兰 郑 华 吴沪生
1 病例资料
1.1 临床资料 女性,12 岁。因“进行性加重的步态异常7年”入首都医科大学附属北京儿童医院(我院)。患儿于7年前无明显诱因出现走路时足尖着地,上楼费力,双上肢活动正常,无发热、关节疼痛症状,到医院就诊,未做特殊治疗,症状逐渐加重;2年前在当地医院行脊柱MRI检查未发现异常,未明确诊断。2年来患儿足尖着地症状逐渐加重,并出现双上肢不能伸直,颈部僵硬,低头受限,无明显四肢无力症状。为进一步诊治来我院就诊,门诊查血乳酸1.6 mmol·L-1,AST 42 U·L-1,CK 866 U·L-1,CK-MB 27 U·L-1;肌电图示肌源性受损,运动和感觉神经传导速度正常。以“肌病待查,多发性关节挛缩症”收入院。
发育史:患儿生后1岁会走,自幼行走较同龄儿慢。智力发育同正常同龄儿。
家族史:其父,39岁,亦有轻度足尖着地史,肘关节及膝关节处肌肉萎缩,1年前因扩张型心肌病去世。父母非近亲结婚。该患儿为第1胎, 足月顺产,独生女。
体格检查: T 37.0℃,P 100·min-1,R 20·min-1,神志清楚,精神反应好;呼吸平稳,心音有力、律齐;腹软,未扪及包块,肝脾肋下均未扪及,肠鸣音正常。面纹对称;眼睑闭合有力,双侧瞳孔等大等圆,对光反射灵敏。双足尖踏地行走(图1A),右侧较重;颈前屈受限,弯腰受限,Schober试验阳性;双侧肘关节挛缩屈曲不能伸直(图1B),肘关节、膝关节近端肌肉挛缩,肘关节远端肥大,双踝关节挛缩,双足背屈受限,足趾活动正常。四肢肌张力正常,双上肢近端肌力Ⅴ级,远端Ⅴ级,双下肢肌力Ⅴ-。双侧肱二头肌、肱三头肌反射均未引出,双侧桡骨膜反射可引出,双侧膝、跟腱反射均未引出。布氏征、克氏征和双侧巴氏征均阴性。
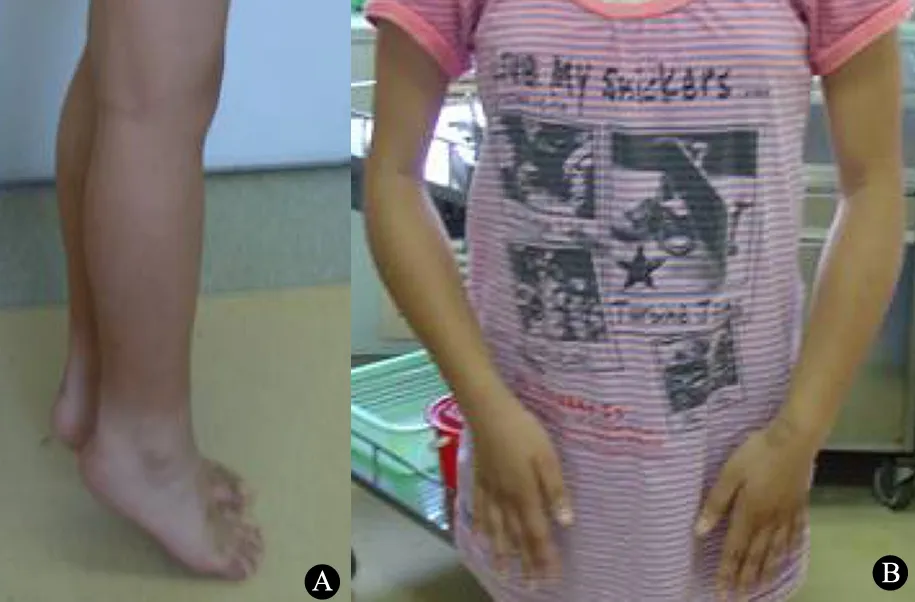
图1 患儿双足尖踏地行走和双侧肘关节挛缩屈曲
Fig 1 Physical performance of EDMD patient
Notes A: Achilles tendon contractures; B: the contracture of the elbows, to 130°
1.2 影像学和实验室检查 动态EEG检查未见异常。肘关节X线正侧位:双肘关节骨质结构未见明显异常,双肘关节近端软组织萎缩。双侧踝关节X线正侧位:双侧踝关节诸骨未见明确骨质异常。超声心动图示心内结构未见明显异常。心肌酶谱:CK 661 U·L-1,CK-MB 33 U·L-1,HBDH 237 U·L-1。
1.3 病理学检查 取左侧股四头肌组织(0.5 cm×1 cm),10%中性甲醛固定,石蜡包埋,切片行苏木精-伊红、改良Gomori、NADA-TR、PAS、油红O和ATPase染色,光镜下可见肌肉细胞大小不均,萎缩和肥大的纤维交替存在, 部分肌纤维代偿性肥大, 脂肪及结缔组织增生明显, 符合肌营养不良改变(图2)。
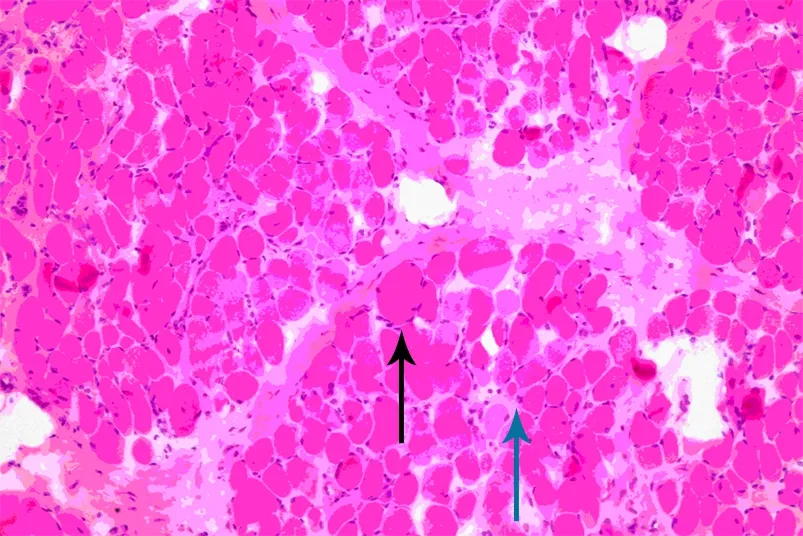
图2 患儿肌肉组织活检光镜下所见(苏木精-伊红染色,×400)
Fig 2 Physical performance of patient(HE,×400)
Notes The pathological changes in muscles showed that the sizes of muscle fibers were different, fiber atrophy(blue arrow) and hypertrophy(black arrow) alternately existed, compensatory hypertrophy of partial muscle fibers existed, fat and connective tissue proliferations were apparent
1.4 基因检测 取患儿外周静脉抗凝血提取基因组DNA,应用PCR 技术进行LMNA基因外显子突变分析,发现LMNA基因外显子4的序列变异c.746G>A(p.Arg249Gln)(图3)。诊断为Emery-Dreifuss肌营养不良症(Emery-Dreifuss muscular dystrophy,EDMD)。查询UMD-LMNA变异数据库,为国外已报道的变异。
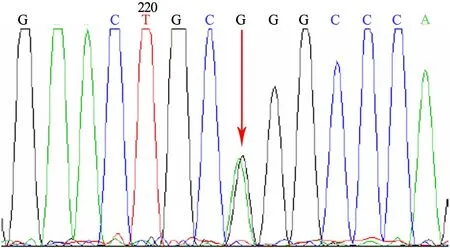
图3 患儿LMNA基因测序结果
Fig 3LMNAgene sequencing
NotesLMNAgene with a heterozygous mutation: c.746G>A(p.Arg249Gln). The arrow pointed the mutation
2 讨论
EDMD是一种儿童期发病的慢性进行性肌营养不良病,绝大多数EDMD为X-连锁隐性遗传(XL-EDMD),少数为常染色体显性遗传(AD-EDMD)和隐性遗传(AR-EDMD),EDMD基因编码蛋白位于细胞核。XL-EDMD基因编码蛋白位于细胞核内面,被命名为Emerin,AD-EDMD基因编码的中间丝蛋白laminA/C位于核浆。1999年确定了AD-EDMD的致病基因是LMNA[1]。Raffaele等[2]在2000年报道了1例AR-EDMD患者。LMNA被确定为AD-EDMD的致病基因[3]。LMNA基因定位于1q21,约24 kb,有12 个外显子。正常情况下,通过选择性剪切拼接,LMNA基因可编码产生4 种A 型核纤层蛋白(A、AΔ10、C、C2),其中核纤层蛋白A 和C 是主要的异构体[2]。XL-EDMD存在Emerin蛋白缺陷,而AD-EDMD表型中Emerin蛋白表达正常,分布与lamins A/C相似,Emerin和lamin A在核内相互作用的功能也许可以解释两者临床表型的相似性。携带LMNA突变的AD-EDMD患者的皮肤成纤维细胞形态学和免疫学分析显示,细胞为蜂窝状胞核结构和核膜水泡样生长,提示lamin A/C的突变可能导致成纤维细胞核膜中支持结构网状系统的减弱,该结构的改变依赖于突变在lamin A/C不同结构域的定位[4]。
位于LMNA基因的氨基端和羧基端的错义突变可导致EDMD,位于该基因中部杆状区域的突变则导致扩张型心肌病,出现传导障碍,而不表现为骨骼肌病变[5]。相关研究表明,LMNA基因突变可引起肢带型肌营养不良症、常染色体隐性Charcot-Marie-Tooth病等多种疾病。单一的LMNA基因突变也可同时引起两种不同的疾病表型[6]。LMNA基因及其编码蛋白lamin A /C异常能引起一组人类遗传病,称为核纤层蛋白病。在所有核纤层蛋白病中,导致常染色体显性遗传的LMNA基因突变位点最多,已经发现约40 种。突变主要为发生在进化过程中比较保守的氨基酸上的错义突变, 也有移码突变和导致翻译提前终止的无义突变。这些突变位点遍布整个基因, 除了个别发生在第12 外显子上外,大多分布在LMNA也就是lamin A/C共有的编码前566个氨基酸的区域。临床表型的严重程度与突变的类型和部位无关。
2010年,国内袁军辉等[7]报道一个AD-EDMD家系先证者及其女儿在LMNA基因第9外显子发现1583(C→G)杂合错义突变,导致第528个氨基酸由苏氨酸替换为精氨酸(T528R)。本文报道的c.746G>A(p.Arg249Gln)变异是在中国人群中首次报道,但该变异在意大利、法国、韩国和日本等均有报道[8~11]。由于患儿父亲有与患儿类似的足尖着地的表现,故考虑其父罹患EDMD的可能性较大,且患儿为LMNA基因突变,符合AD-EDMD的诊断。但患儿父亲已死亡,故无法对其进行基因分析,该患儿也无其他兄弟姐妹,其所知的父母双方亲属及上一代均无类似临床表现者,故未进行家系分析。
本文所报道的变异c.746G>A(p.Arg249Gln)能够引起AD-EDMD及肢带型肌营养不良症两种不同表型[10,12]。肢带型肌营养不良症发病年龄在20~30岁,少数在10岁左右或中年;呈常染色体隐性或显性遗传,但大部分为散发病例。首先累及肩胛带或骨盆带肌肉,从上肢扩展到下肢或从下肢扩展到上肢,有腓肠肌或其他肌肉的肥大。疾病严重程度和进展速度差别较大,通常在出现症状20~30年后丧失运动能力。骨骼畸形和关节挛缩少见,即使出现也在晚期。
EDMD常在儿童期发病, 是一种相对良性的肌营养不良类型,病情进展缓慢,临床特征包括①早期出现肢体关节挛缩;②缓慢进展的肢体肌肉萎缩与无力;③心肌与心脏传导改变。与其他类型肌营养不良所不同的是EDMD在肌肉出现明显无力和萎缩之前,肘、跟腱和脊柱关节出现挛缩,相继出现四肢无力,以双侧肱二头肌、肱三头肌、胫前肌和腓肠肌尤为明显。肌肉无力分布呈肱-腓分布, 即分别累及上肢近端和下肢远端, 随病情进展,后期肌无力可扩展到肢带肌近端,如肩胛肌和骨盆带肌肉。肌无力在30岁前进展较缓慢,30岁后进展加快,AD-EDMD患者丧失步行能力较常见,在XL-EDMD患者少见。EDMD的严重程度和进展程度差异很大,即使是相同的基因突变类型,发病年龄、肌肉和心脏受累的严重程度在不同家系间或同一家系成员间也存在着显著差异[13]。本例患儿的临床特征表现为在肌肉出现明显无力和萎缩之前,肘、跟腱和脊柱关节出现挛缩。肌肉无力分布呈肱-腓分布,尚未累及肩胛带和骨盆带肌肉,符合EDMD的临床特征。
本例患儿12岁,未发现心肌受累的证据,这与Benedetti等[14]研究一致,在>25岁的EDMD患者,心脏受累的发生率为76%;<25岁的患者,心脏受累的发生率仅为10%;在儿童期发病的患儿,几乎仅表现为骨骼肌受累。由于AD-EDMD患者能表达正常数量的核纤层蛋白A/C,虽能通过免疫学方法检测核纤层蛋白A/C,但不能作为诊断依据,因此基因检测是确诊AD-EDMD的最可靠方法[15,16]。心脏受累是EDMD最严重的并发症。AD-EDMD患者发生室性快速心律失常和扩张型心肌病的概率高于XL-EDMD患者,某些患者可见广泛性心肌病,导致猝死或发展为进行性心功能衰竭,约半数患者可发生猝死。一旦发现心脏传导阻滞,应尽早安装起搏器,延长生命。故建议对进行性双侧对称的肌无力伴肘关节、跟腱挛缩和脊柱僵硬患者均应行心功能检查和LMNA基因分析,以期对EDMD早期诊断,可有效减少猝死发生。
EDMD患者肌肉病理学表现为非特异性改变,可见肌纤维大小不等、肌核增加、结缔组织增多、肌纤维坏死和巨噬细胞增多等。多数患者电镜下超微结构可见肌核结构的异常,包括形状不规则和(或)染色质的重组,并有核周和核内的空泡出现,染色质的改变也出现在50%的肌卫星细胞。本例患儿的肌肉病理改变与文献报道相符[17],由于肌肉活检病理学检查缺乏诊断特异性改变。故EDMD的临床诊断主要依靠其临床表现。
本病需与Ullrich型先天性肌营养不良症(Ullrich congenital muscular dystrophy,UCMD)鉴别。UCMD是一种较为常见的先天性肌营养不良,一般在出生前或生后不久起病,临床特征为近端关节挛缩与远端关节活动度过大同时存在,患儿常因关节挛缩丧失行走能力。随着疾病进展, 远端关节弹性过度被肌肉挛缩掩盖。还可有一些特殊表现如跟骨后凸、先天性髋关节脱位和斜颈等,智力正常[18]。EDMD发病年龄较晚,在肌肉出现明显无力和萎缩之前,肘、跟腱和脊柱关节即出现挛缩,可与之鉴别。EDMD还需与Duchenne型肌营养不良症、强直性脊柱炎等疾病相鉴别。Duchenne型肌营养不良症也可出现关节挛缩,但多发生在晚期肌肉无力导致废用性关节挛缩,且主要累及踝、膝和髋关节,之后波及肘关节。强直性脊柱炎为一种慢性、进行性炎症疾病,病变部位主要累及骶髂关节、脊椎、脊椎旁软组织和四肢关节,常有关节疼痛和肿胀,同时可见ESR和CRP升高、HLA B27阳性等实验室检查指标的异常。
[1]Bonne G, Di Barletta MR, Varnous S, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet, 1999, 21(3): 285-288
[2]Di Barletta MR, Ricci E, Galluzzi G, et al . Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am J Hum Gene, 2000, 66(4): 1407-1412
[3]Ostlund C, Worman HJ. Nuclear envelope proteins and neuromuscular diseases. Muscle Nerve, 2003, 27(4): 393-406
[4]Mercuri E, Poppe M, Quinlivan R, et al. Extreme variability of phenotype in patients with an identical missense mutation in the lamin A/C gene: from congenital onset with severe phenotype to milder classic Emery-Dreifuss variant. Arch Neurol, 2004, 61(5):690-694
[5]Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med, 1999, 341(23):1759-1762
[6]Walter MC, Witt TN, Weigel BS, et al. Deletion of the LMNA initiator codon leading to a neurogenic variant of autosomal dominant Emery-Dreifuss muscular dystrophy. Neuromuscul Disord, 2005, 15(1): 40-44
[7]Yuan JH(袁军辉), Hu J, Zhao Z, et al. Mutation analysis of a Chinese family with autosomal dominant Emery-Dreifuss muscular dystrophy. Chin J Med Genet(中华医学遗传学杂志), 2010, 27(2):136-139
[8]Boriani G, Gallina M, Merlini L, et al. Clinical relevance of atrial fibrillation/flutter, stroke, pacemaker implant, and heart failure in Emery-Dreifuss muscular dystrophy: a long-term longitudinal study.Stroke, 2003, 34(4):901-908
[9]Deconinck N, Dion E, Ben Yaou R, et al. Differentiating Emery-Dreifuss muscular dystrophy and collagen VI-related myopathies using a specific CT scanner pattern. Neuromuscul Disord, 2010, 20(8):517-523
[10]Ki CS, Hong JS, Jeong GY, et al. Identification of lamin A /C (LMNA) gene mutations in Korean patients with autosomal dominant Emery-Dreifuss muscular dystrophy and limb-girdle muscular dystrophy 1B. J Hum Genet, 2002, 47(5):225-228
[11]Astejada MN, Goto K, Nagano A, et al. Emerinopathy and laminopathy clinical, pathological and molecular features of muscular dystrophy with nuclear envelopathy in Japan. Acta Myol, 2007, 26(3):159-164
[12]Rudenskaya GE, Polyakov AV, Tverskaya SM, et al. Laminopathies in Russian families.Clin Genet, 2008, 74(2): 127-133
[13]Higuchi Y, Hongou M, Ozawa K, et al. A family of Emery-Dreifuss muscular dystrophy with extreme difference in severity. Pediatr Neurol, 2005, 32(5): 358-360
[14]Benedetti S, Menditto I, Degano M, et al. Phenotypic clustering of lamin A/C mutations in neuromuscular patients. Neurology, 2007, 69(12):1285-1292
[15]Colomer J, Iturriaga C, Bonne G, et al. Autosomal dominant Emery-Dreifuss muscular dystrophy: a new family with late diagnosis. Neuromuscul Disord, 2002, 12(1):19-25
[16]Lassuthová P, Baránková L, Kraus J, et al. Emery-Dreifuss muscular dystrophy: a novel mutation in the LMNA gene. Pediatr Neurol, 2009, 41(2):127-130
[17]Park YE, Hayashi YK, Goto K, et al. Nuclear changes in skeletal muscle extend to satellite cells in autosomal dominant Emery-Dreifuss muscular dystrophy/limb-girdle muscular dystrophy 1B. Neuromuscul Disord, 2009, 19(1):29-36
[18]Zhu WH(朱雯华), Zhao CB, Lu JH, et al. Clinical and pathological features in 3 Chinese patients with Ullrich congenital muscular dystrophy. Chin J Neurol(中华神经科杂志), 2008, 41(8): 536-540
