用于染料敏化太阳能电池的D5同类物分子设计
2010-12-12詹卫伸李源作陈茂笃
詹卫伸 潘 石 李源作 陈茂笃
(大连理工大学物理与光电工程学院,近场光学与纳米技术研究所,辽宁大连 116023)
Since Grätzel et al.[1]reported the dye-sensitized solar cells (DSSC)based on Ru complex in 1991,more attention has been paid to DSSC due to its comparatively low cost and high efficiency[1-3].However,DSSC sensitized by free-metal organic dye has attracted more interests from researchers because of its much lower cost[4-11].In particular,the structural diversity and the simple synthetic route of organic dye molecules provide conditions for seeking more competitive DSSC sensitizer.Thus,the molecular design of organic dyes comes into development to improve the function of DSSC[12-20].
The dye molecules applied to DSSC should have a structure of“donor-conjugate π bridge-acceptor(D-π-A)”[5,19,21-25],in which the“electron acceptor”must contain the“anchoring group”.By the“anchoring group”,dye molecules can absorb onto the surface of TiO2nanocrystalline[6,13,15,20,25-26].At present,the“anchoring group”is usually chosen as carboxyl(—COOH).Most D-π-A dyes take dialkylamines or diphenylamine moieties as electron donor,carboxylic acid,cyanoacrylic acid or rhodanine-3-acetic acid moieties as electron acceptor which also acts as an anchoring group.Carboxy groups can hang on the surface of TiO2,providing a strong restriction to dyes and good electron-channel. Photoabsorption characteristic of D-π-A dyes is connected with intramolecular charge transfer(ICT)excitation from electron donor to electron acceptor moiety,which results in efficient electron transfer from dyes excitation state via electron acceptor moiety (carboxy groups)to TiO2conduction band edge(CB).Charge transfer or separation between electron donors and acceptors in the excitation state can facilitate the electron injection from dyes to TiO2CB,thus it can separate the cationic charge from surface and effectively prevent the photoelectron(the injected electron) from compounding with oxidized dyes[12,25].
Thelowestunoccupiedmolecularorbital(LUMO)energy level positions of dye molecules and their absorption spectra are the most key factors for the application of dyes to DSSC[27].LUMO energy level of dye must be higher than TiO2conduction band edge,and the higher it is,the larger the driving force for electron injection from dyes excitation state to TiO2will be,which is more beneficial to the improvement of DSSC[10,17-18,22,26,28-31]. Nowadays,the good match between UV-Vis absorption spectra of synthetical sensitized DSSC dyes and that of the solar radiation spectra has not been achieved,which is one of the urgent tasks.It is reported that,the lengthening of conjugated bridge of dye molecular structure could lead to red-shift in molecular absorption spectra[16,20,32].But the length should be proper,as overlong conjugated bridge will cause the accumulation of dye molecules on the surface of TiO2,affecting the properties of DSSC[4,8,25].
Generally,the abilities of electron donor group(D)donating electrons,electron acceptor group(A)accepting electrons and the electric properties of conjugate π bridge have great influence on the photophysical,electrochemical and intramolecular chargetransfer properties of D-π-A dyes.The need of high-efficiency DSSC can be met by the change of chemical structure of every element(D,A,or conjugate π bridge).Absorption spectra of dye molecules should match solar radiation spectra,and their LUMO and HOMO energy levels must match the conduction band edge energy level of TiO2electrode and the redox potential of I-/I-3electrolyte.For study on the properties of molecule in photophysics and photochemistry,the molecular nonadiabatic process has attracted much attention in recent years.Han et al.[33-35]has investigated the nonadiabatic phenomenon of small molecules in detail.For larger moecules,however,nowadays we can not carry out accurate research on nonadiabatic process and we do not discuss this issue particularly in this paper.
Recently,an organic dye called D5(Fig.1)attracts more attention because it can sensitize DSSC to enhance its solar energy conversion efficiency by 5%,and are expected to be applied in solid DSSC[7,15,23,26,36-40].But the wavelength of D5 at maximum absorbance is 476 nm,and the maximum wavelength of D5 absorbed on the surface of TiO2blue-shifts to 444 nm[41],which se-riously affects the ability of dye to absorb solar energy.Organic dye D5 is not considered as the bestphotosencitizer.Itispossible to find a better photo-sencitizer used for DSSC by modifying D5 molecules.
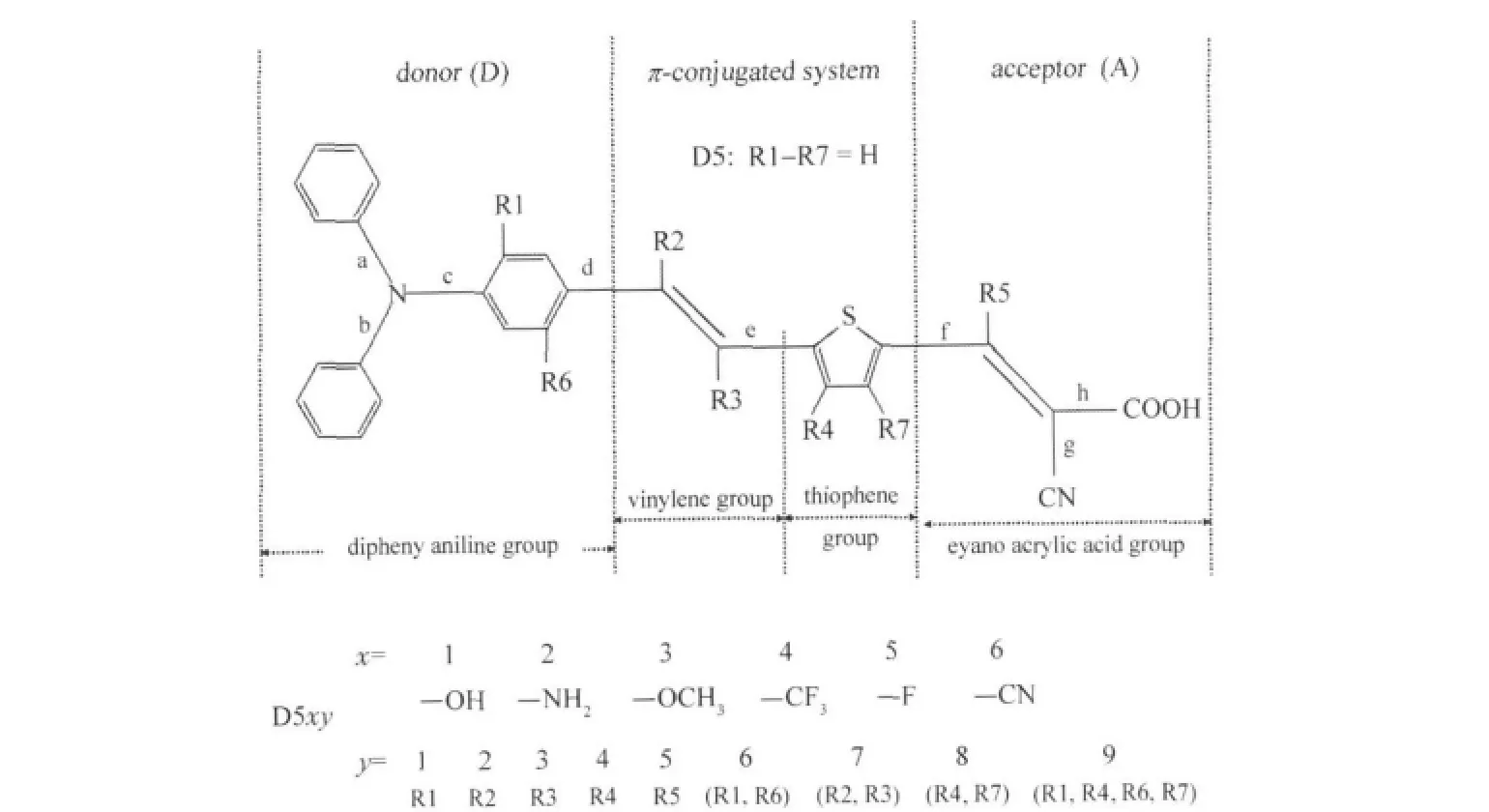
Fig.1 Molecular structures of dye D5 and its analogues(D5xy)x and y in D5xy denote different substituents and positions based on D5.For example,D536,x=3,y=6,using—OCH3to replace—H in the positions of R1 and R6.
Before trying any synthesis,the density functional theory (DFT)[42-43]and time-dependent DFT(TDDFT)[44-47]were adopted to provide a sound theoretical foundation for the design and filtering of analogues according to their molecular energy levels and absorption spectra[9,14,16,29-30,36,48-51].Based on D5,an attempt on the design of more efficient dye molecules was made via calculation.During the design,the principle of designing a much simpler dye molecule was insisted on.The method of modifying molecules is to introduce substitutent group to D5 skeleton.
1 Computational methods
DFT and TDDFT calculations were performed using the Gaussian 03 software package[52].The ground-state geometries of D5 and its analogues were fully optimized in vacuum without any symmetry constraints at the B3LYP level of theory with the 6-31G(d)basis set.The contribution of singly excited state configurations to each electronic transition and the simulated absorption spectra of the D5 analogues were calculated.The electronic absorption spectra require calculation of the allowed excitations and oscillator strengths.These calculations were carried out using TDDFT with the same basis set and exchange-correlation functional in vacuum and solution.The TD-B3LYP calculation containing the solvation effect in ethanol was performed on the geometries optimized in vacuum.DFT and TDDFT with B3LYP functional calculations qualitatively coincide with the experiment results[30,40-41,53-55].The conductor polarizable continuum model(CPCM)[24,56]is conducted using parameters and iterative computation methods suggested by Klamt[57-58]to contain the solvation effect.Natural bond orbital(NBO)analysis was performed in order to analyze the charge populations of the D5 analogues[40,59].
2 Results and discussion
2.1 Molecular design of D5 analogues
For the dye of sensitized DSSC,the energy levels of HOMO and LUMO and its absorption spectra are the most important properties.To obtain efficient electron injection from excited dye to the conduction band of the TiO2electrode,the LUMO energy level of dye molecule must be higher than the conduction band energy level of the TiO2.Not every excited dye molecule can inject electrons to TiO2electrode,because of many other processes causing the de-excitation of dye molecule,which has a strong impact on the electron injection to the TiO2electrode. But with higher energy level of the LUMO,the driving force for the electron injection from dye molecule to TiO2electrode will become stronger,which results in the higher transfer efficiency of DSSC.In order to make the oxidized(lose electrons)dye molecule efficiently recover(gain electrons)from I-/I-3redox couple in the electrolyte,the HOMO energy level of dye molecule has to be lower than the redox potential of I-/I-3.To gain higher lightharvesting efficiency,dye molecule must have greater molar absorption coefficient in the wide area of solar spectra.Solar radiation spectra at the area of 500-600 nm is the strongest.If the absorption spectra of sensitized DSSC dye molecule are included in this region,it will be a perfect sensibilizer.However,up to now,there is not any kind of organic dye molecule suited for DSSC,whose absorption spectra can match perfectly with the solar radiation spectra.Especially,the absorption spectra of dye molecule on TiO2electrode exhibits significant blue-shift,compared with that in the vacuum and polar solvent.So far,the absorption spectra of dye molecule are mainly in the shortwave zone of near ultraviolet region and visible region.Therefore,the red-shift of absorption spectra is still the primary standard to judge the quality of sensitized DSSC dye molecule.The D5 type dye molecule for DSSC will be designed according to the HOMO and LUMO energy levels calculated by DFT and the maximum wavelength of the absorption spectra calculated by TDDFT.
The results of experiment and calculation demonstrate that D5 is a better free-metal organic dye for DSSC[23,26,36,38,40-41,60].Because of the variety of organic molecular structure,it is possible to design organic dye superior to D5 for DSSC by modifying D5 molecule.Based on our previous investigation[27],the modification to D5 should make the new D5 molecule maximize LUMO energy level,on the condition that its HOMO energy level is lower than I-/I-3,so that the driving force for electron injection of the dye molecule from the excitation state to TiO2electrode could become greater,which can raise conversion efficiency of DSSC. Besides,dye molecular absorption spectra should be red-shifted as much as possible,in order to make the absorption of solar radiation photons more efficient.
The extension of conjugate π bridge in dye molecular structure and the introduction of the electron-donating substituent and electron-withdrawing substituent to the chromophore backbone could move the HOMO and LUMO energy levels of dye molecules and also cause the red shift in absorption spectrum, which provide a possibility for the adjustments of the photochemical and electrochemical properties of dyes.It should be noticed that the expansion of conjugated π bridge in D-π-A dye molecules can easily lead to the accumulation of dye molecules on TiO2surface,reducing electron-injection yields from the dye molecules to the TiO2conduction band because of intermolecular energy transfer between molecules.In the modification of D5 analogue dye molecules,we did not extend the length of the conjugate π bridge.In addition,the geometry of the dye molecule should not be too large;otherwise,the dye molecules adsorbed onto TiO2surface per unit area as well as the conversion efficiency of DSSC per unit area will be reduced.
As shown in Fig.1,taking—H from the skeleton of D5 molecule in different positions,we modified the D5 molecules with the electron-donating substituent(—OH,—NH2,—OCH3) and the electron-withdrawing substituent(—CF3,—F,—CN),respectively.Xia et al.[30]have used this method to modify the cou-marin molecule.To facilitate discussion,we numbered the modified organic molecules(Fig.1).We have designed 54 kinds of molecules,6 of which(D521,D526,D529,D531,D544,D545) failed in the DFT optimization.
By DFT/TDDFT calculation(see Sections 2.3 and 2.4),it is shown that D516,D536 and D537,not only induce red-shift in absorption spectrum relative to D5,but also make LUMO energy level be greater than that of D5.The DSSC sensitized by these types of organic molecules make use of solar energy more efficiently and enlarge the driving force for electron injection into the TiO2electrode.Moreover,its HOMO energy level is lower than the position of the redox I-/I-3.Therefore,the conversion efficiency of the DSSC adopting these types of organic molecules as sensitizers should be greater than that of D5.It should be noted that these types of organic molecules share some common characteristics:firstly,the substituents are all the electron-donating substituent,symmetrically being replaced in pair;secondly,the substituents are located in the electron-donating group or in the conjugate bridge near the electron-donating group.The LUMO energy level position of D516 is the highest,so the driving force for electron injection from excited state into the TiO2is the greatest.
DFT/TDDFT calculation results(see Sections 2.3 and 2.4) show that the LUMO energy levels of D565,D567 and D568 are lower than that of D5(much higher than conduction band edge of TiO2),the driving force for electron injection from excited state of these molecules into the TiO2is lower than that of D5,which leads to the lower electron transfer rate compared with D5,but their absorption spectra are red-shifted intensely compared with D5,making full use of solar energy.And their HOMO energy levels are lower than that of redox I-/I-3.Therefore,the conversion efficiency of the DSSC adopting these types of molecules as sensitizers is higher than that of D5.These types of organic molecules also share some common characteristics:the substituents are all the electron-withdrawing substituent;the substituents may be located in the electron-donating group,the electron-accepting group or the conjugate bridge.Therefore,the introduction of the electron-withdrawing substituent into D5 dye molecules may cause strong red-shift in the absorption spectra of organic molecules.
DFT/TDDFT calculation results of other substituted molecules indicate that,compared with the DFT/TDDFT calculations of D5, either LUMO energy level has been improved,with blue-shift in absorption spectrum,or absorption spectrum is red-shifted,with lowerLUMO energy levels.
2.2 Molecular structures
Theconjugateπbridgeofnon-planeisnotconductivetoICTof electron transition from the electron-donating group to the electron-accepting group[11,14-16].Fig.2 shows the optimized molecular structures of D5 and some of its analogues.The conjugate bridges of all the D5 analogues except for D537 are all plane, which is just the same as D5.
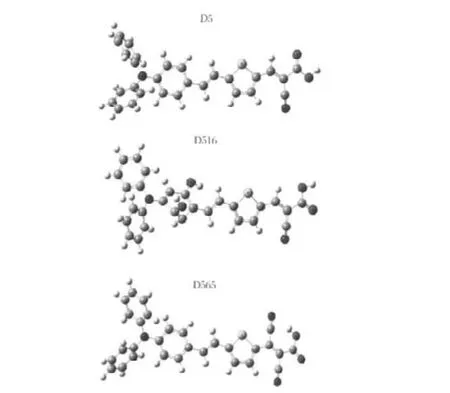
Fig.2 Optimized geometrical structures of dye D5 and its analogues
By analysis of NBO,we find that the charge populations of donor group,conjugated bridge,and acceptor group of D5 are 0.061e,0.114e,and-0.174e,respectively,which is a typical D-π-A molecular structure.For D536,they are 0.057e,0.126e,and -0.184e;for D537,they are 0.057e,0.130e,and-0.184e;for D565,they are 0.097e,0.184e,and-0.281e;for D567,they are 0.184e,-0.094e,and-0.092e;for D568,they are 0.130e,-0.021e, and-0.105e.All of these molecules have similar D-π-A structures.The charge populations of three groups of D516 are 0.154e, 0.043e,and-0.197e,respectively.The positive charges of the electron-donating group and the negative charges of electronaccepting group of D516 are much more than that of D5,but the charges of conjugate π bridge are lower than that of D5.Therefore,this analogue should have D-π-A molecular structure superior to D5.
Table 1 lists part of the bond lengths of chemical bonds of D5 and its analogues,from which we can find their difference is lower than 1 pm in general,and the greatest difference among individuals is lower than 3 pm.Therefore,it is believed that the molecular stabilities of D5 and its analogues are on the whole the same.
The electronic structures of HOMOs and LUMOs of D5 and its analogues D516 and D565 are shown in Fig.3.The HOMOs of D5 are π orbitals,while LUMOs are single states of π*orbi-tals.HOMOs have ground-state characteristics,while LUMOs have excited state characteristics.In the ground state,the electrons are mainly distributed in the electron donor(diphenyl anilinegroup)andconjugated bridge(vinylene and thiophene group). In the excited state,the electrons are distributed in the thiophenes and cyanoacrylic acid groups,but mostly in the anchoring group(carboxyl:—COOH).For D5 analogues,HOMOs(HOMO and HOMO-1)are π orbital which is located in the electron donor groups,electron acceptor groups and the conjugate bridge, or the whole molecule.And the LUMOs(LUMO and LUMO+1) are π*orbital which is located in the conjugate bridge and the electron acceptor(anchor)group.The electronic orbitals of D5 analogues are the same as that of D5 due to their similar molecularstructures.Underillumination,throughintramolecularcharge transfer,electrons move from HOMOs to LUMOs,and eventually reach the anchoring groups.In this system,the light-induced electronic distribution change is considered as one of the decisive factors resulting in efficient charge separation.Thus it can be concluded:the electron transport from electron donor to electron acceptor(anchor)occurred during the course of excitation of dye molecules.As dye molecules are adsorbed on the semiconductor surface via the carboxyl group,the excitation promotes the electron injection process.The electronic structures of D5 and its analogues are very beneficial to solar cells.
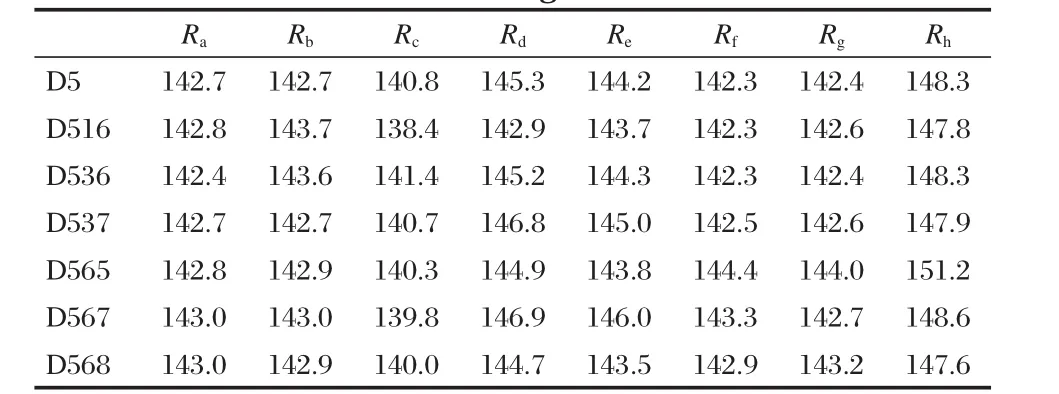
Table 1 Selected bond length (R,in pm)of the dye D5 and its analogues

Fig.3 Isodensity plots(isodensity contour=0.02 a.u.)of the frontier orbitals of dye D5 and its analogues
2.3 Energy level diagram
The diagram for calculated frontier molecular orbital energy levels of the D5,D516,and D565 in vacuum and ethanol is depicted in Fig.4.The LUMO energy level positions of D536, D537,D565,and D567 in vacuum are-2.585,-2.558,-3.374, and-3.374 eV(in ethanol:-2.667,-2.667,-3.347,and-3.320 eV),respectively.The HOMO energy level positions of D536, D537,D565,andD567invacuumare-4.980,-5.061,-5.361,and-5.551 eV(in ethanol:-4.898,-4.980,-5.061,and-5.306 eV), respectively.Whether in vacuum or in the polar solution,LUMO energy level of D5 was much higher than that of TiO2conduction band edge(ca-4.0 eV).Thus,D5 molecule,that is still in excitation state after absorbing photons could inject electrons to TiO2electrode smoothly.HOMOs of D5 are all lower than that of I-/I-3(ca-4.8 eV),therefore,D5 molecule that loses electrons could be restored by getting electrons from electrolyte.

Fig.4 The frontier molecular orbital energy levels of dye D5 and its analoguesH-1:HOMO-1,H-2:HOMO-2,L+1:LUMO+1,L+2:LUMO+2
LUMO energy level positions of D516,D536,and D537 are all higher than those of D5,thus,the driving forces for electron injection from molecular excitation state of D516,D536 and D537 to TiO2electrode are larger than that of D5.LUMO energy level positions of D565,D567,and D568 are all lower than that of D5,but higher than that of TiO2conduction band edge; molecules in excited states of D565,D567,and D568 have a strong ability to inject electrons into TiO2electrodes.
HOMO energy level positions of D5 and its analogues are all lower than that of I-/I-3,therefore,D5 and its analogues,which lose electrons,could be restored by getting electrons from electrolyte.Overall,the positions of LUMOs energy level(LUMO especially)decline in solution,but those of HOMOs(HOMO especially)have slight increases,and LUMOs-HOMOs gaps decrease,causing red-shift in the molecular absorption spectra in solution.
The HOMO-LUMO gap of the dye D5 and analogues in vacuum was 1.987-2.503 eV(in ethanol:1.714-2.285 eV).Among D5,D516,D536,and D537,HOMO-LUMO gaps of D516 and D536 are the smallest.Among D565,D567,and D568,D565 has the smallest HOMO-LUMO gap.
Table 2 shows the energy level positions of D5 in vacuum and different solutions.In different polar solutions,MO energies are almost the same.It is clear that different polar solutions have the same effect on MOs energies.Compared with the case of vacuum,the polar solutions have little effect on LUMO+1 and HOMO-1 of D5,and almost no effect on LUMO,but comparatively great effect on HOMO energy level,which could enhance HOMO energy level by about 0.1 eV.Polar solutions decrease the HOMO-LUMO energy gap of D5,which is the main reason for red-shift in dye molecular absorption spectrum.From the HOMO and LUMO energy levels in vacuum and solution,it can conclude that dye molecules in polar solution produce solvent effects of red-shift in absorption spectra[40,61],whose physical mechanism is that the polar solution has no effect on the LUMO energy level of dye molecules,but can improve the HOMO energy level of dye molecules.
2.4 Electronic absorption spectra
Fig.5 shows the UV-Vis absorption spectra of D5 and ana-logues D516 and D565 by calculation.Obviously,in the UV-Vis region,the absorption of long wave is more intense than that of the short one.There is a clear red-shift in the absorption spectra of D5 molecule in the solution compared with that in the vacuum,which is aforenamed solvent effect.In ethanol,MeCN,and THF solution,the absorption spectra of D5 are almost the same, namely,the solvent effects of different solutions are nearly identical due to the same impact of the aforementioned polar solution on the MOs energy level of the dye molecules.In the UVVis region,D5 has two clear absorption bands.In vacuum,the central wavelength of the first absorption band is 537 nm,and that of the second one is 393 nm.Because of solvent effect,in the polar solution,the central wavelengths of the two absorption bands of D5 have a red-shift to 603 nm and 415 nm.While the wavelengths of the two absorption peaks acquired by experiment in the solution were 476 nm and 300 nm,respectively[41]. Compared with the experiment,the calculation of the TDDFT shows a considerable red-shift,especially the red-shift in the solution is more visible than the one in vacuum.The difference between the experiment and calculation of the TDDFT may come from two aspects:the calculation method and solvent effect.The energy gaps calculated by the DFT are always smaller than that of the factual one,especially for the bigger conjugated system, which causes low calculated excited energy and significant redshiftinthecalculatedabsorptionspectraandthefactual one[26,40,62]. The solution,especially the polar solution(such as MeCN and THF),through the long-range interaction between solute and solvent,influences the geometry,the electronic structure,and the properties of molecule.Thus,solvent effect causes the energy level of solute molecule to decrease,thereby making a significant red-shift in the absorption band.It shows that it is difficult to conform the calculation to the experiment quantitatively. Though there is difference,the calculation of TDDFT can still describe the spectral features of the D5,because the line shape and the relative intensity of spectra correspond with the experiment qualitatively.
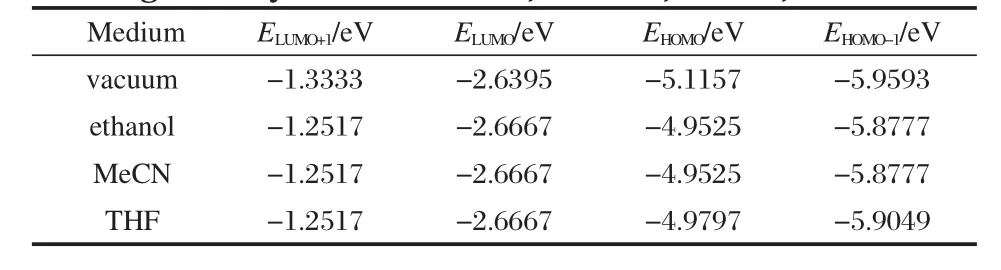
Table 2Calculated LUMO+1,LUMO,HOMO,and HOMO-1 energies of dye D5 in vacuum,ethanol,MeCN,and THF
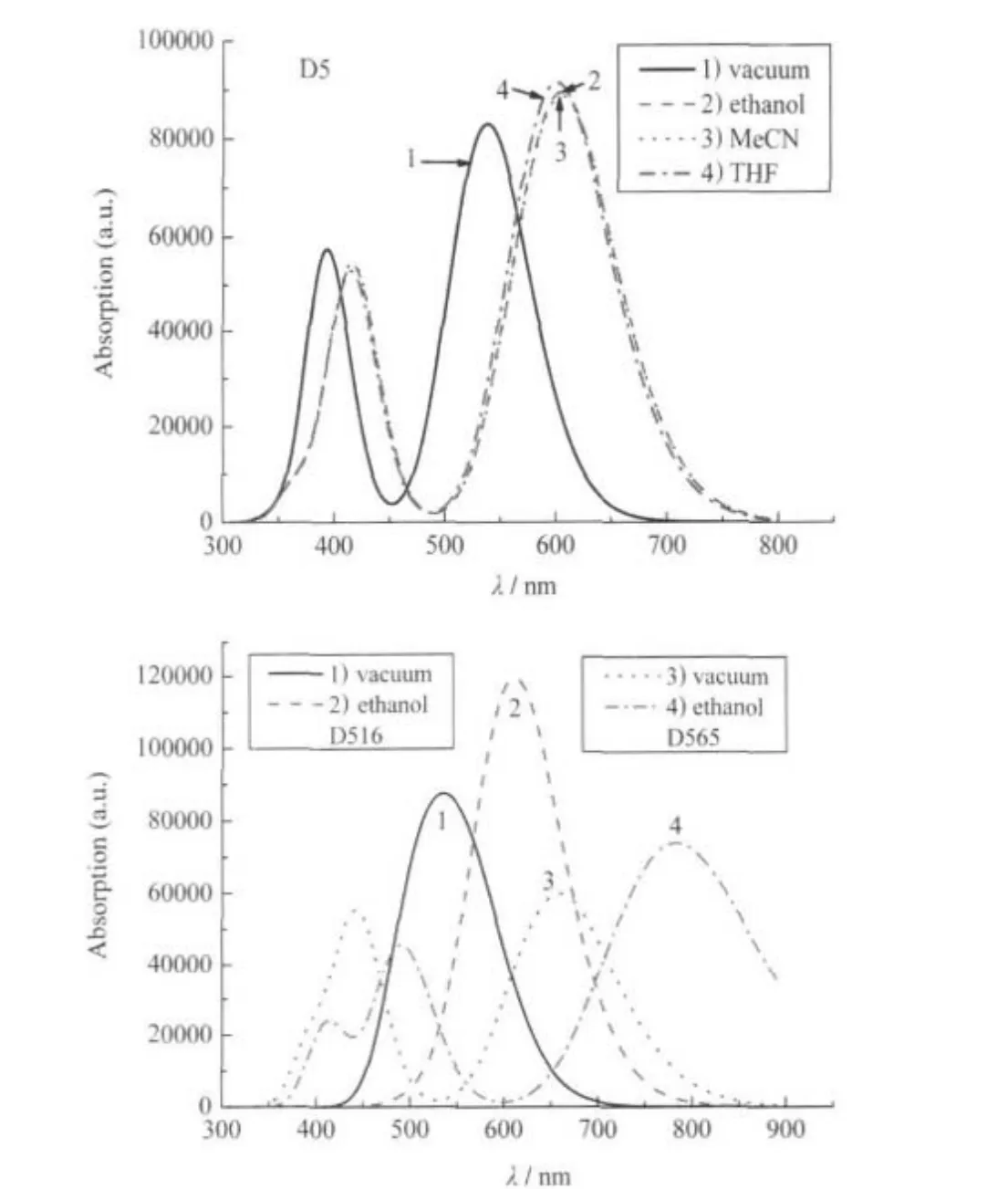
Fig.5 Calculated electronic absorption spectra of dye D5 and its analogues in different media
The absorption peaks of D516,D536,and D537 in vacuum were 562,560,and 562 nm,respectively,and those in alcoholic solution were 611,627,and 634 nm,respectively.Compared with the absorption spectra of D5(in vacuum and alcoholic solution,the absorption peaks λmaxwere 537 and 603 nm),there are significant red-shifts in the absorption spectra of these D5 analogue molecules.But the molar absorption coefficient ε(31000 and 32000 a.u.in vacuum and alcohol solution,respectively)of D537 at absorption peak are less than those of D516(83000 and 120000 a.u.in vacuum and in alcohol solution,respectively)and D536(70000 and 78000 a.u.in vacuum and in alcohol solution, respectively).The redshifts in absorption spectra of D516 and D536 are basically the same relative to D5.The molar absorption coefficient of D516 at absorption peak is greater than that of D536;thus,D516 is a sensitizer superior to D537 and D536.
The absorption peaks of D565,D567,and D568 in vacuum were 658,616,and 622 nm,respectively,and the ones in alcohol solution were 782,702,and 719 nm,respectively,with intense red-shift relative to D5 and the same maximum molar absorption coefficient as that of D5.In terms of the absorption spectrum,they are better than D5 as sensitizer.D565 had the largest red-shift.Considering that the calculation of TDFDT itself may cause red-shift in the absorption spectra,and that the molecular absorption on TiO2could cause blueshift,D565 absorption spectrum is promising to match solar UV-Vis spectra better.D565 is a DSSC sensitizer superior to D567 and D568.
In order to know the microscopic information about the absorption bands and electronic transitions,the relevant MO properties should be studied.Since visible and near-UV region are significant spectrum regions for photo-to-current conversion, Table 3 shows the singlet-singlet transitions in the absorption bands of D5 and its analogues.Simulated spectra about the absorption bands near 603 nm in solution shows that initial and final states of electronic transitions mostly lie in HOMO and LUMO, respectively.The initial and final states of electronic transitions mostly lie in HOMO-1 and LUMO respectively in the absorption bands near 415 nm.Weak absorption bands are also detected (353 nm in vacuum,367 nm in solution correspondingly).For these transitions,initial states are the two former HOMOs,and final states are the two former LUMOs.According to the analysis of electronic transitions and molecular orbitals,three absorption bands(two of which in visible region)in UV-Vis region for D5 molecules are typical π→π*transitions.
Either in vacuum or in solution,D5 and its similar molecules show similar characters that the strongest absorption band (which is also the largest absorption band of red shift)is mainly composed of initial state HOMO and final state LUMO,while the other absorption bands are mainly composed of the(HOMO-2)-HOMO to the LUMO-(LUMO+2).These transitions caused by absorption are π→π*transitions.In these transitions,initial states are mainly related to the molecular orbitals of electrondonor groups,while the final states are mainly related to the molecular orbitals of electron acceptor groups.This shows that absorption is photoinduced electron transfer process.Thus,the excitations generate charge separated states.
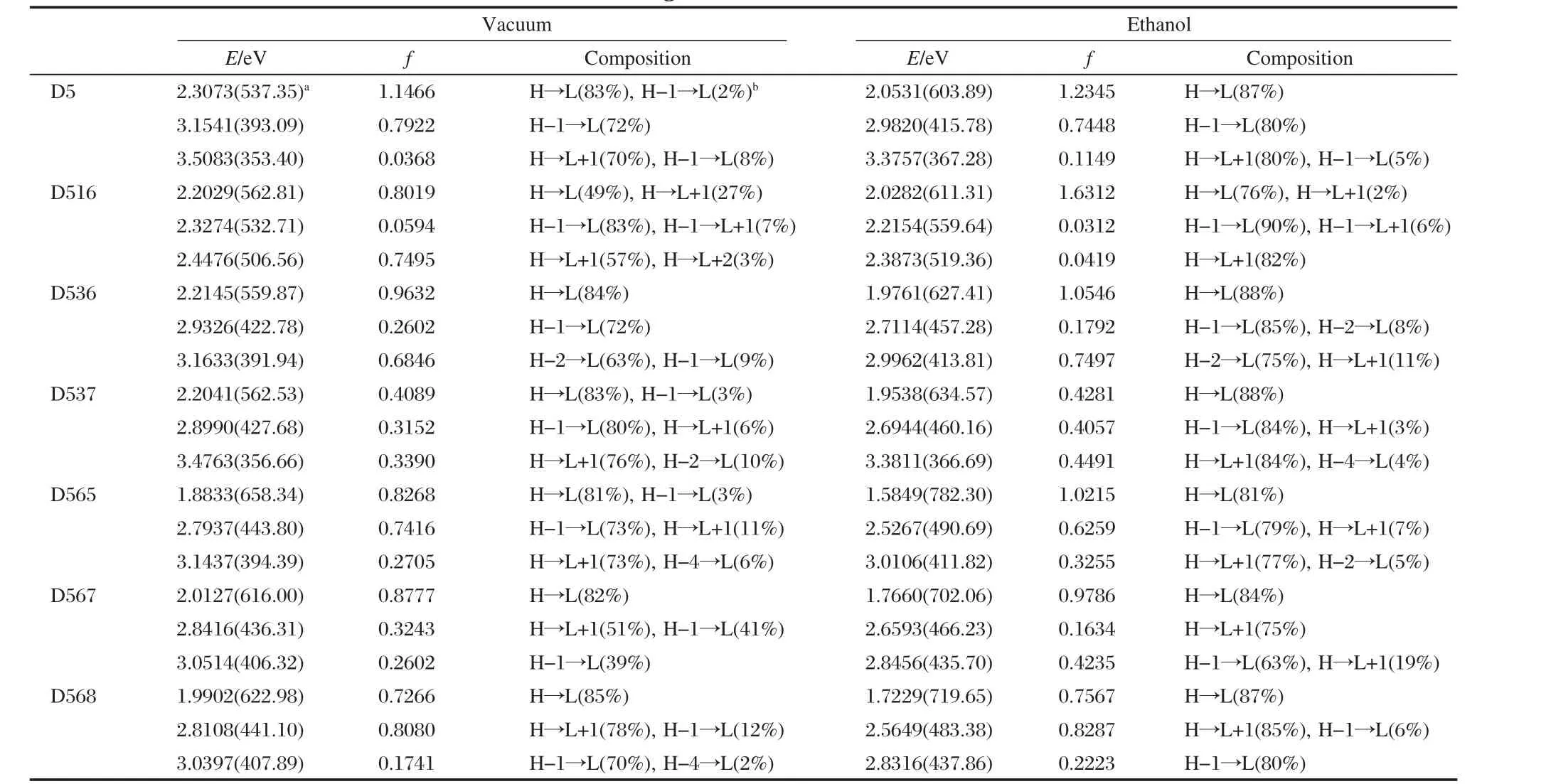
Table 3 Computed excited energies(E),oscillator strengths(f)and two highest electronic transition configurations for dye D5 and its analogues in vacuum and ethanol
D516′s energy level gaps(LUMO)-(LUMO+1)and(LUMO+ 1)-(LUMO+2)were 0.626 and 0.626 eV,respectively(0.816 and 0.599 eV in the solution,respectively),which are the smallest in the D5 and its analogues.This indicates that,the density of states in D516 is more plentiful than other elements in the vicinity of LUMO,which is the main reason why D516 has the greatest molar absorption coefficient at absorption peak in the visible region.
3 Conclusions
Considering both the molecular orbital energy(HOMO and LUMO energy levels)and the absorption spectra,LUMO energy levelsofD5analoguemolecules(D516,D536,D537)areallhigher than that of D5,the driving forces for electron injection from excitation state to TiO2electrode are larger;the absorption spectra of such dye molecules all red-shift compared with D5. Therefore,D516,D536,and D537 are DSSC sensitizers superior to D5.Further analysis shows that,among the D5 analogues, with the greatest molar absorption coefficient,D516 is the best DSSC sensitizer.
For absorption spectra,the absorption spectra of D5 analogue molecules(D565,D567,D568)all intensely red-shift compared with D5,which could capture solar radiation photons more efficiently than D5,enhancing the utilization efficiency of solar energy.Among them,the spectra of D565 red-shift most strongly, with the greatest molar absorption coefficient,which could match the solar spectra better after being absorbed onto the surface of TiO2.
With the assistance of NBO analysis,the designed D5 analogue molecules D516,D536,D537,D565,D567,and D568, which are promising to be superior to D5,all share D-π-A structure,among which conjugation bridges are of planar structure,which benefits the generation of charge separated state from dye molecules by optical excitation.The analysis of DFT electron structure shows that,HOMOs of these molecules lie on π orbitals of electron donor,and LUMOs lie on π*orbitals of electron acceptor.TDDFT analysis on the excitation energy(absorption spectroscopy)shows that,the optical excitation causes π-π*transition,resulting in intramolecular charge transfer,with electronic absorption spectra lying in the zone from near-ultraviolet to visible light.
In summery,it is possible to design free-metal organic dye molecules applied to DSSC using DFT/TDDFT.Calculated results show that,the designed molecules D516 and D565 are expected to be free-metal organic dye molecules applied to DSSC superior to D5.
1 O′Regan,B.;Grätzel,M.Nature,1991,353:737
2 Grätzel,M.Inorg.Chem.,2005,44:6841
3 Peter,L.M.Phys.Chem.Chem.Phys.,2007,9:2630
4 Wang,Z.S.;Cui,Y.;Dan-oh,Y.;Kasada,C.;Shinpo,A.;Hara,K. J.Phys.Chem.C,2007,111:7224
5 Chen,R.;Yang,X.;Tian,H.;Sun,L.C.J.Photochem.Photobiol. A-Chem.,2007,189:295
6 Tian,H.;Yang,X.;Chen,R.;Pan,Y.;Li,L.;Hagfeldt,A.;Sun,L. C.Chem.Commun.,2007:3741
7 Kim,S.;Kim,D.;Choi,H.;Kang,M.S.;Song,K.;Kang,S.O.; Ko,J.Chem.Commun.,2008:4951
8 Ito,S.;Miura,H.;Uchida,S.;Takata,M.;Sumioka,K.;Liska,P.; Comte,P.;Péchy,P.;Grätzel,M.Chem.Commun.,2008:5194
9 Li,C.;Yum,J.H.;Moon,S.J.;Herrmann,A.;Eickemeyer,F.; Pschirer,N.G.;Erk,P.;Schöneboom,J.;Müllen,K.;Grätzel,M.; Nazeeruddin,M.K.ChemSusChem,2008,1:615
10 Jin,Y.;Hua,J.;Wu,W.;Ma,X.;Meng,F.Synth.Met.,2008,158: 64
11 Burke,A.;Ito,S.;Snaith,H.;Bach,U.;Kwiatkowski,J.;Grätzel, M.Nano Lett.,2008,8:977
12 Chen,Z.;Li,F.;Huang,C.Curr.Org.Chem.,2007,11:1241
13 Rochford,J.;Chu,D.;Hagfeldt,A.;Galoppini,E.J.Am.Chem. Soc.,2007,129:4655
14 Tsai,M.S.;Hsu,Y.C.;Lin,J.T.;Chen,H.C.;Hsu,C.P.J.Phys. Chem.C,2007,111:18785
15 Chen,R.;Yang,X.;Tian,H.;Wang,X.;Hagfeldt,A.;Sun,L.C. Chem.Mater.,2007,19:4007
16 Choi,H.;Lee,J.K.;Song,K.H.;Song,K.;Kang,S.O.;Ko,J. Tetrahedron,2007,63:1553
17 Park,J.K.;Lee,H.R.;Chen,J.;Shinokubo,H.;Osuka,A.;Kim,D. J.Phys.Chem.C,2008,112:16691
18 Eu,S.;Hayashi,S.;Umeyama,T.;Matano,Y.;Araki,Y.;Imahori, H.J.Phys.Chem.C,2008,112:4396
19 Yen,Y.S.;Hsu,Y.C.;Lin,J.T.;Chang,C.W.;Hsu,C.P.;Yin,D. J.J.Phys.Chem.C,2008,112:12557
20 Li,G.;Jiang,K.J.;Li,Y.F.;Li,S.L.;Yang,L.M.J.Phys.Chem. C,2008,112:11591
21 Hagberg,D.P.;Marinado,T.;Karlsson,K.M.;Nonomura,K.;Qin, P.;Boschloo,G.;Brinck,T.;Hagfeldt,A.;Sun,L.C.J.Org. Chem.,2007,72:9550
22 Qin,P.;Yang,X.;Chen,R.;Sun,L.C.;Marinado,T.;Edvinsson, T.;Boschloo,G.;Hagfeldt,A.J.Phys.Chem.C,2007,111:1853
23 Boschloo,G.;Marinado,T.;Nonomura,K.;Edvinsson,T.;Agrios, A.G.;Hagberg,D.P.;Sun,L.C.;Quintana,M.;Karthikeyan,C.S.; Thelakkat,M.;Hagfeldt,A.Thin Solid Films,2008,516:7214
24 Balanay,M.P.;Kim,D.H.Phys.Chem.Chem.Phys.,2008,10: 5121
25 Ooyama,Y.;Harima,Y.Eur.J.Org.Chem.,2009:2903
26 Marinado,T.;Hagberg,D.P.;Hedlund,M.;Edvinsson,T.; Johansson,E.M.J.;Boschloo,G.;Rensmo,H.;Brinck,T.;Sun,L. C.;Hagfeldty,A.Phys.Chem.Chem.Phys.,2009,11:133
27 Zhan,W.S.;Pan,S.;Li,Y.Z.;Chen,M.D.Acta Phys.-Chim.Sin., 2009,25:2087 [詹卫伸,潘 石,李源作,陈茂笃.物理化学学报,2009,25:2087]
28 Gervaldo,M.;Fungo,F.;Durantini,E.N.;Silber,J.J.;Sereno,L.; Otero,L.J.Phys.Chem.B,2005,109:20953
29 Zhang,X.H.;Li,C.;Wang,W.B.;Cheng,X.X.;Wang,X.S.; Zhang,B.W.J.Mater.Chem.,2007,17:642
30 Zhang,X.;Zhang,J.J.;Xia,Y.Y.J.Photochem.Photobiol.AChem.,2008,194:167
31 Xu,W.;Peng,B.;Chen,J.;Liang,M.;Cai,F.J.Phys.Chem.C, 2008,112:874
32 Sayama,K.;Tsukagoshi,S.;Mori,T.;Hara,K.;Ohga,Y.;Shinpo, A.;Abe,Y.;Suga,S.;Arakawa,H.Sol.Energy Mater.Sol.Cells, 2003,80:47
33 Chu,T.S.;Zhang,Y.;Han,K.L.Int.Rev.Phys.Chem.,2006,25: 201
34 Chu,T.S.;Varandas,A.J.C.;Han,K.L.Chemical Physics Letters, 2009,471:222
35 Chu,T.S.;Han,K.L.;Hanke,M.;Balint-Kurti,G.G.; Kuppermann,A.;Abrol,R.J.Chem.Phys.,2009,130:144301
36 Hagberg,D.P.;Yum,J.H.;Lee,H.;Angelis,F.D.;Marinado,T.; Karlsson,K.M.;Humphry-Baker,R.;Sun,L.C.;Hagfeldt,A.; Grätzel,M.;Nazeeruddin,M.K.J.Am.Chem.Soc.,2008,130: 6259
37 Chou,C.S.;Yang,R.Y.;Weng,M.H.;Yeh,C.H.Powder Technology,2008,187:181
38 Agrios,A.G.;Hagfeldt,A.J.Phys.Chem.C,2008,112:10021
39 Fredin,K.;Johansson,E.M.J.;Blom,T.;Hedlund,M.;Plogmaker, S.;Siegbahn,H.;Leifer,K.;Rensmo,H.Synth.Met.,2009,159: 166
40 Zhang,C.R.;Liu,Z.J.;Chen,Y.H.;Chen,H.S.;Wu,Y.Z.;Yuan, L.H.J.Mol.Struct.-Theochem,2009,899:86
41 Hagberg,D.P.;Edvinsson,T.;Marinado,T.;Boschloo,G.; Hagfeldt,A.;Sun,L.C.Chem.Commun.,2006:2245
42 Hohenberg,P.;Kohn,W.Phys.Rev.,1964,136:B864
43 Kohn,W.;Sham,L.J.Phys.Rev.,1965,140:A1133
44 Bauernschmitt,R.;Ahlrichs,R.Chemical Physics Letters,1996, 256:454
45 Wiberg,K.B.;Stratmann,R.E.;Frisch,M.J.Chemical Physics Letters,1998,297:60
46 Hirata,S.;Head-Gordon,M.Chemical Physics Letters,1999,302: 375
47 Bauernschmitt,R.;Haiser,M.;Treutler,O.;Ahlrichs,R.Chemical Physics Letters,1997,264:573
48 Kim,S.;Lee,J.K.;Kang,S.O.;Ko,J.;Yum,J.H.;Frantacci,S.; Angelis,F.D.;Censo,D.D.;Nazeeruddin,M.K.;Grätzel,M. J.Am.Chem.Soc.,2006,128:16701
49 Jacquemin,D.;Perpète,E.A.;Scuseria,G.E.;Ciofini,I.;Adamo, C.J.Chem.Theory Comput.,2008,4:123
50 Wonga,B.M.;Cordaro,J.G.J.Chem.Phys.,2008,129:214703
51 Di Censo,D.;Fantacci,S.;De Angelis,F.;Klein,C.;Evans,N.; Kalyanasundaram,K.;Bolink,H.J.;Grazel,M.;Nazeeruddin,M. K.Inorg.Chem.,2008,47:980
52 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision C.02.Pittsburgh,PA:Gaussian Inc.,2003
53 Zhao,G.J.;Han,K.L.ChemPhysChem,2008,9:1842
54 Zhao,G.J.;Han,K.L.J.Phys.Chem.A,2007,111:2469
55 Zhao,G.J.;Han,K.L.J.Phys.Chem.A,2009,113:14329
56 Barone,V.;Cossi,M.J.Phys.Chem.A,1998,102:1995
57 Klamt,A.J.Phys.Chem.,1995,99(7):2224
58 Klamt,A.J.Phys.Chem.,1996,100(9):3349
59 Reed,A.E.;Weinstock,R.B.;Weinhold,F.J.Chem.Phys.,1985, 83:735
60 Quintana,M.;Marinado,T.;Nonomura,K.;Boschloo,G.;Hagfeldt, A.J.Photochem.Photobiol.A-Chem.,2009,202:159
61 Cossi,M.;Rega,N.;Scalmani,G.;Barone,V.J.Comput.Chem., 2003,24:669
62 Wang,Y.L.;Wu,G.S.Acta Phys.-Chim.Sin.,2007,23:1831 [王溢磊,吴国是.物理化学学报,2007,23:1831]
