六核钽铑二元混合簇合物的几何和电子结构
2010-10-14林梦海
曹 飞 谭 凯 林梦海
(1九江学院化学化工学院,江西九江 332005; 2厦门大学化学化工学院化学系,福建厦门 361005)
六核钽铑二元混合簇合物的几何和电子结构
曹 飞1,2,*谭 凯2林梦海2
(1九江学院化学化工学院,江西九江 332005;2厦门大学化学化工学院化学系,福建厦门 361005)
采用密度泛函理论对六核钽、铑八面体纯簇及其混合簇的几何结构和电子性质进行了研究.计算结果表明:大部分钽铑混合簇稳定构型的对称性均较低,为C1或Cs点群,只有[Ta2Rh4Cl4H8(CN)6]4-团簇的稳定构型对称性较高,为C2h或C4v点群;混合簇的最高占据分子轨道(HOMO)与最低未占据分子轨道(LUMO)能隙(ΔEH-L)均较小,介于0.52-1.00 eV之间;混合簇的前线轨道主要由骨架金属原子的d电子贡献,随着Rh原子替代Ta原子个数的递增,Ta—Rh键对混合簇稳定构型所起作用逐渐增加,Ta—Ta键所起作用减小,而Rh—Rh键为非键或反键性质.
密度泛函理论; 过渡金属团簇; 金属键; 钽; 铑
Abstract:Density functional theory(DFT)was employed to investigate the geometric and electronic properties of a series of hexanuclear binary Ta/Rh clusters.The results show that most of the stable structures of the Ta/Rh mixed clusters have low symmetry and belong to the C1or Cspoint groups while the[Ta2Rh4Cl4H8(CN)6]4-clusters are highly symmetric(C2hor C4v).The energy gaps(ΔEH-L)between the highest occupied molecular orbital(HOMO)and the lowest unoccupied molecular orbital(LUMO)of the mixed clusters are narrow and within 0.52-1.00 eV.A frontier orbital analysis of the mixed clusters clearly shows that the molecular orbitals are mainly composed of the d-orbitals of the skeleton metal atoms.When more rhodium atoms are substituted for tantalum atoms,the Ta—Rh bond has a greater influence on the stable structures of the mixed clusters and the influence of Ta—Ta bond decreases while the Rh—Rh bond has a non-bonding or anti-bonding nature.
Key Words: Density functional theory;Transition-metal cluster;Metal bond;Tantalum;Rhodium
一般来说,六核过渡金属原子簇化合物可以分为两类[1].第一类团簇由前过渡金属元素与π-给予配体(如Cl)组成.例如八面体团簇[Nb6Cl12(CN)6]4-,其价电子数为76,最近几年的研究[2-9]表明,它可作为组装开放式框架材料的亚结构单元.第二类是由低氧化态的后过渡金属与π-接受配体(如CO)组成,如八面体团簇Rh6(CO)16,其价电子数为86.2004年,Weller小组[10]合成的簇合物[Rh6(PR3)6H12]2+,虽然由后过渡金属元素Rh构成,但其分子构型和电子结构类似于前过渡金属团簇,这将为前后过渡金属团簇的研究架起一座桥梁.该纯铑簇合物能在常温常压的条件下可逆地加氢和释放氢,是理想的储氢材料[11-13].目前国内外有关六核过渡金属簇合物的研究主要集中在纯簇上[14-18],混合团簇配位化合物的理论研究鲜有报道[19-21].因而本文将应用密度泛函理论,通过对该系列混合簇的稳定构型、电子结构、成键分子轨道组成和Mulliken布居进行分析,探索钽铑二元混合簇的化学键和电子结构的递变规律,希望对指导设计和合成新的前后过渡金属簇提供理论依据.
1 计算方法
计算采用基于密度泛函理论的DMol3程序[22],电子交换关联能函数用局域密度近似(LDA)的Vosko-Wilk-Nusair(VWN)泛函,采用双数值基加极化函数(DNP)基组,对Ta、Rh原子采用有效核赝势(ECP)处理,总能收敛判据为10-6a.u.,几何优化用Broyden-Fletcher-Goldfarb-Shanno(BFGS)方法,结构优化以其梯度、位移和能量是否收敛为判据,精度分别优于2×10-3Ry·(a.u.)-1、5×10-3和 1×10-5a.u..在优化构型的基础上做了频率分析,以保证得到的结构为稳定构型.
2 结果与讨论
2.1 单金属六核钽、铑八面体簇合物
根据张乾二提出的多面体分子轨道理论的群论方法[23],可推测[(M6X12)L6]4-(M=Nb,Ta)簇合物骨架的电子结构为M:(A1g)2(T1u)6(T2g)6(A2u)2.根据Brayshaw[13]和Lin[15]等人的研究结果可知,[Rh6H12(CN)6]4-团簇中12个H原子(配体提供的价电子数μ=1)替代[Ta6Cl12(CN)6]4-团簇中12 个 Cl原子(μ=3),电子数减少了24;但同时6个Rh原子(d9)较6个Ta原子(d5)刚好多出24个电子,故这两种团簇的价电子数均为76.[Ta6Cl12(CN)6]4-团簇骨架成键分子轨道刚好排满,最低未占据分子轨道(LUMO)为反键轨道,则其最高占据分子轨道(HOMO)与LUMO的能隙(ΔEH-L)应较大.[Rh6H12(CN)6]4-团簇骨架 Rh6多出的24个电子应排在反键或非键轨道上,d电子数增加,轨道能量相差不大,ΔEH-L应较小.
计算结果显示(见图1),[Ta6Cl12(CN)6]4-的最外层是由8个Ta—Ta成键分子轨道占据,其电子排布为(A1g)2(T1u)6(T2g)6(A2u)2.[Rh6H12(CN)6]4-的最外层则是由12个Rh—Rh非键分子轨道占据,其能级排布为(T2u)6(T2g)6(Eu)4(T1g)6(A2g)2.[Ta6Cl12(CN)6]4-和[Rh6H12(CN)6]4-构型对称性均为Oh(见图2),自旋为零.Ta—Ta键长为0.293 nm,Rh—Rh键长为0.271 nm,Ta和Rh原子的Mulliken电荷(Q)分别为0.06e和-0.78e.HOMO与LUMO的能隙(ΔEH-L)分别为1.48和0.37 eV,与文献值[13,24]非常吻合.
2.2 六核二元钽、铑混合簇的几何构型
为确保钽铑混合团簇的价电子数为76,当用1个Rh原子替代[Ta6Cl12(CN)6]4-团簇中1个Ta原子时,骨架多出4个电子,故需要同时用2个H(μ=1)原子配体替代2个Cl(μ=3)原子,从而得到一系列价电子数为76的二元钽铑混合簇:[Ta5RhCl10H2(CN)6]4-、[Ta4Rh2Cl8H4(CN)6]4-、[Ta3Rh3Cl6H6(CN)6]4-、[Ta2Rh4Cl4H8(CN)6]4-、[TaRh5Cl2H10(CN)6]4-.
2.2.1 [Ta5RhCl10H2(CN)6]4-团簇
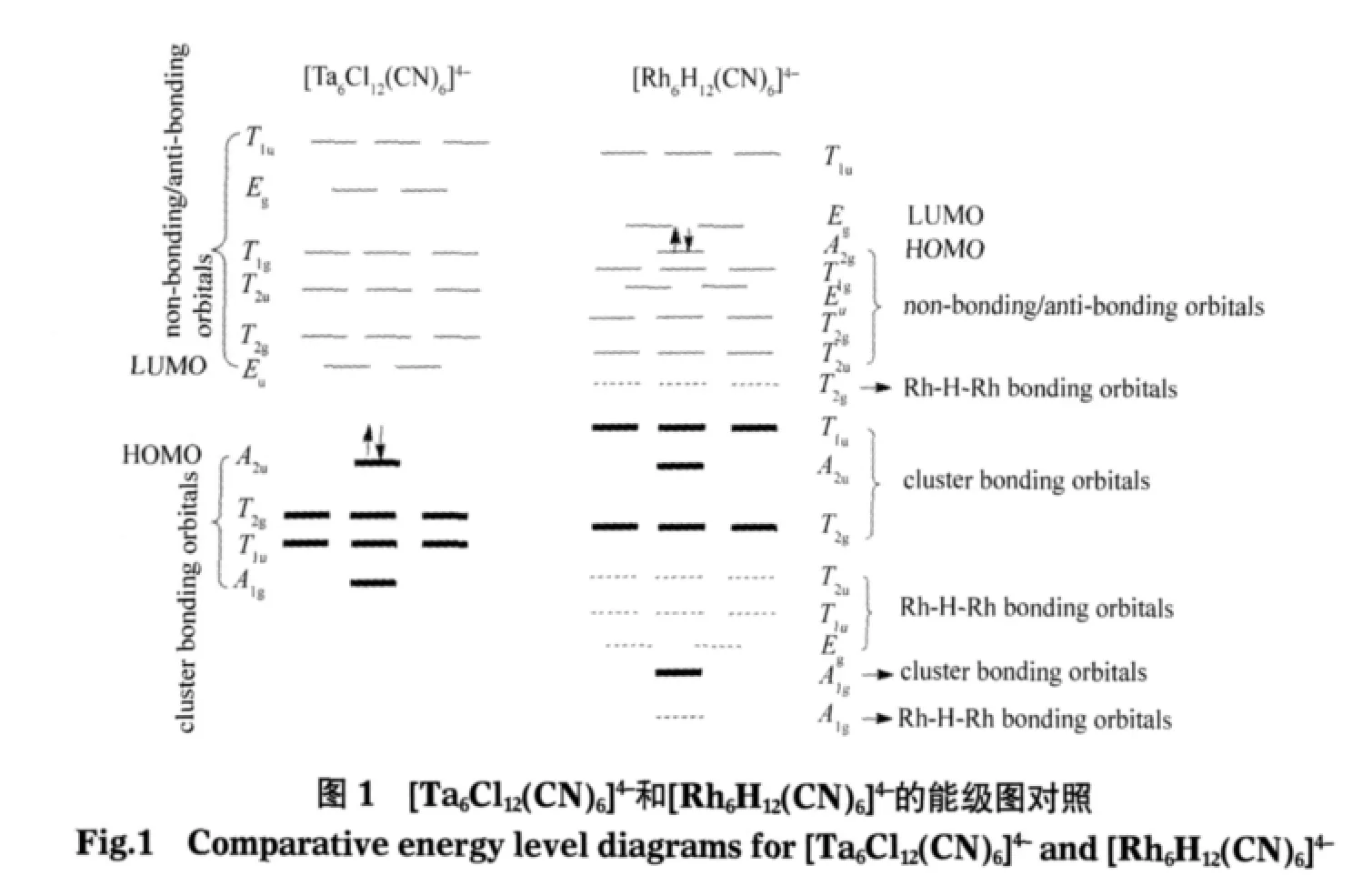
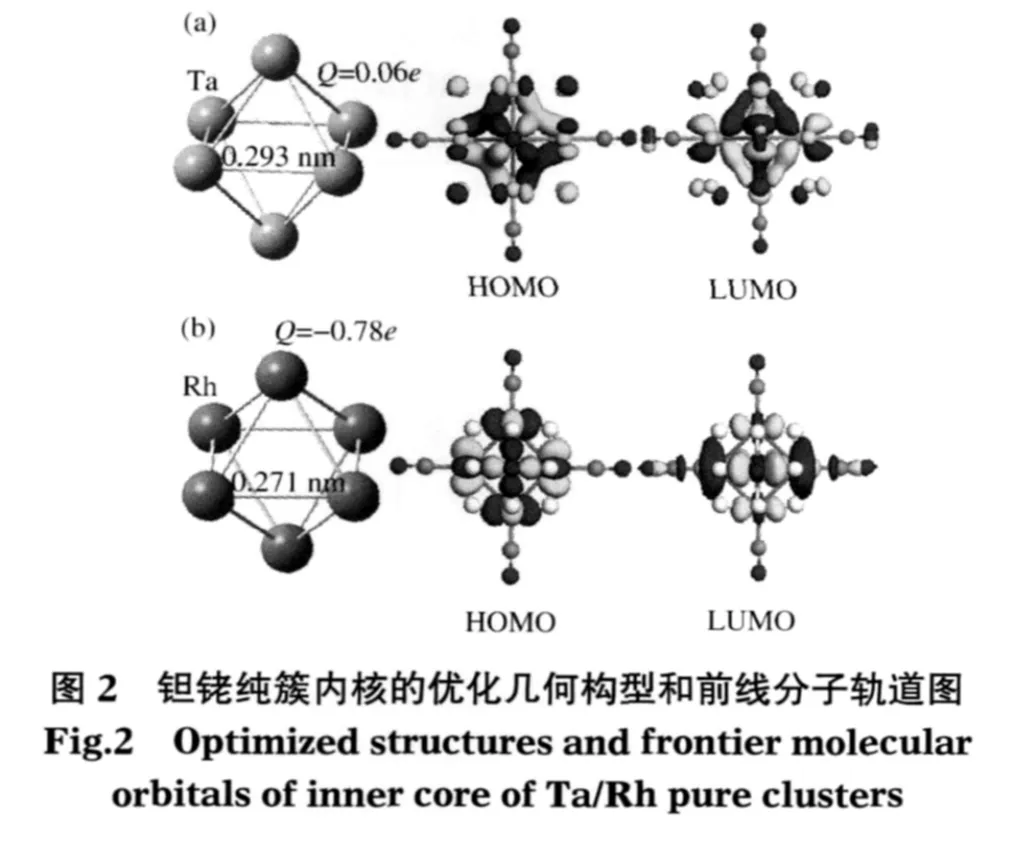
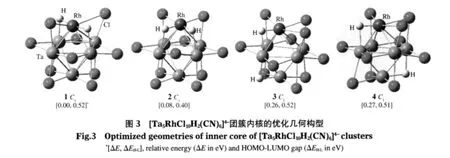
图3列出了4种能量较低[Ta5RhCl10H2(CN)6]4-团簇的稳定构型.计算证实,以2个H原子位于Rh原子对位时的结构(构型1)为最稳定构型,邻位时(构型2)次之.H原子离Rh原子较远的异构体3和4的能量较高.构型1中Ta—Ta键长相对于[Ta6Cl12(CN)6]4-中的Ta—Ta键长变化不大,介于0.287-0.320 nm之间,Ta—Rh键长介于0.264-0.305 nm之间(见表1).
2.2.2 [Ta4Rh2Cl8H4(CN)6]4-团簇
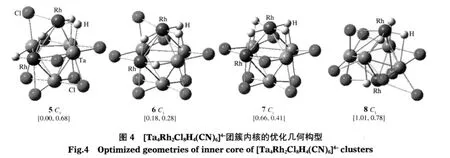
我们列出4个能量较低的稳定异构体(见图4).在这些异构体中,内核Ta4Rh2的Rh原子处于邻位时的构型5能量最低,而当Rh原子处于对位时(构型8)在能量上比最稳定构型5高1.01 eV.构型5-7含5个Ta—Ta键,构型8含4个Ta—Ta键,可见含Ta—Ta键较多的构型稳定.计算证实对称性高的异构体能量相对较高,有的甚至不稳定(有虚频).构型5中Ta—Ta键长为0.283-0.312 nm.Rh—Ta键长为0.261-0.291 nm(见表1).
2.2.3 [Ta3Rh3Cl6H6(CN)6]4-团簇
图5列出9个计算得到能量较低的无虚频稳定构型.在这些异构体中,Rh原子两两相邻的构型(9-11)能量相对较低,其它构型(12-17)能量相对较高.同[Ta4Rh2Cl8H4(CN)6]4-混合簇相似,也是含Ta—Ta键较多的构型9-11稳定(表2).对于配体,H原子易与Rh原子配位,且H之间彼此相邻的构型(9-11)能量较低;若H原子比较分散(构型16-17),则能量较高.计算也证实对称性高的异构体能量相对较高,甚至不稳定.最稳定构型9中的Ta—Ta键长相对于构型5有所增长,为0.295-0.304 nm.Rh—Ta键长为0.254-0.278 nm,较构型5中Rh—Ta键长有所减小(表1).
2.2.4 [Ta2Rh4Cl4H8(CN)6]4-团簇
通过频率计算,列出4个能量较低的稳定构型(图6).与前面两种Ta/Rh混合团簇不同,含Ta—Rh键较多的构型(18-20)能量较低,构型相对稳定;含Ta—Rh键较少的构型21能量较高(表2).而且配体对称分布的对称性较高的构型(18为C2h群,19为C4v群)较稳定,构型19的能量仅比构型18高0.07 eV.最稳定构型18的HOMO与LUMO能隙(ΔEH-L=0.65 eV)较小(见表3),具备作为储氢材料的条件.
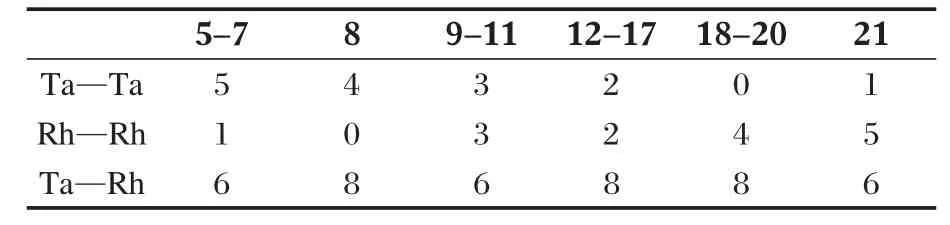
表2Ta/Rh混合簇中M—M(M=Ta,Rh)键的个数Table 2 The number of M—M(M=Ta,Rh)bond for the Ta/Rh mixed clusters
2.2.5 [TaRh5Cl2H10(CN)6]4-团簇
图7列出了计算得到的4个[TaRh5Cl2H10(CN)6]4-团簇的稳定构型.计算证实此类二元混合簇相对于正八面体变形很大,且多数构型不稳定(有虚频).在这些异构体中,最稳定构型22中配体Cl原子相距较远,能量最低.次稳定结构23中Cl原子位于邻位.Cl原子位于对位时的构型不稳定.构型22中的Rh—Rh键长为 0.266-0.366 nm,Rh—Ta键长为0.239-0.272 nm(表1).
2.3 成键和电子性质

从[Ta5RhCl10H2(CN)6]4-到[TaRh5Cl2H10(CN)6]4-混合 团 簇 中,最 稳 定 的 构 型 1、5、9、18 和 22 的HOMO与LUMO间的能隙(ΔEH-L)及Ta、Rh原子的Mulliken电荷如表3所示.随着Rh原子替代Ta原子个数的递增,Ta的原子Mulliken电荷逐渐增大(0.15e→0.44e),Rh原子的Mulliken电荷为负值,有变小的趋势(-0.79e→-0.88e),但变化不大.可见内核电子逐渐由Ta原子转移到Rh原子.
除构型1外,从构型5到构型22中,Ta—Ta键的Mulliken布局(MBO)有减小趋势(0.12e→0.10e),键长逐渐增长(0.294 nm→0.299 nm),成键能力减弱.Ta—Rh键的Mulliken布局有增加趋势(0.16e→0.20e),键长有逐渐减小趋势 (0.279 nm→0.262 nm),成键能力增强.可见随着Rh原子替代Ta原子个数的递增,对于稳定构型Ta—Ta键所起的作用减少,而Ta—Rh键所起的作用增加.计算证实,从[Ta4Rh2Cl8H4(CN)6]4-到[Ta2Rh4Cl4H8(CN)6]4-的稳定构型,由含Ta—Ta键个数多的构型5、9稳定,变化到Ta—Rh键个数多的构型18稳定.
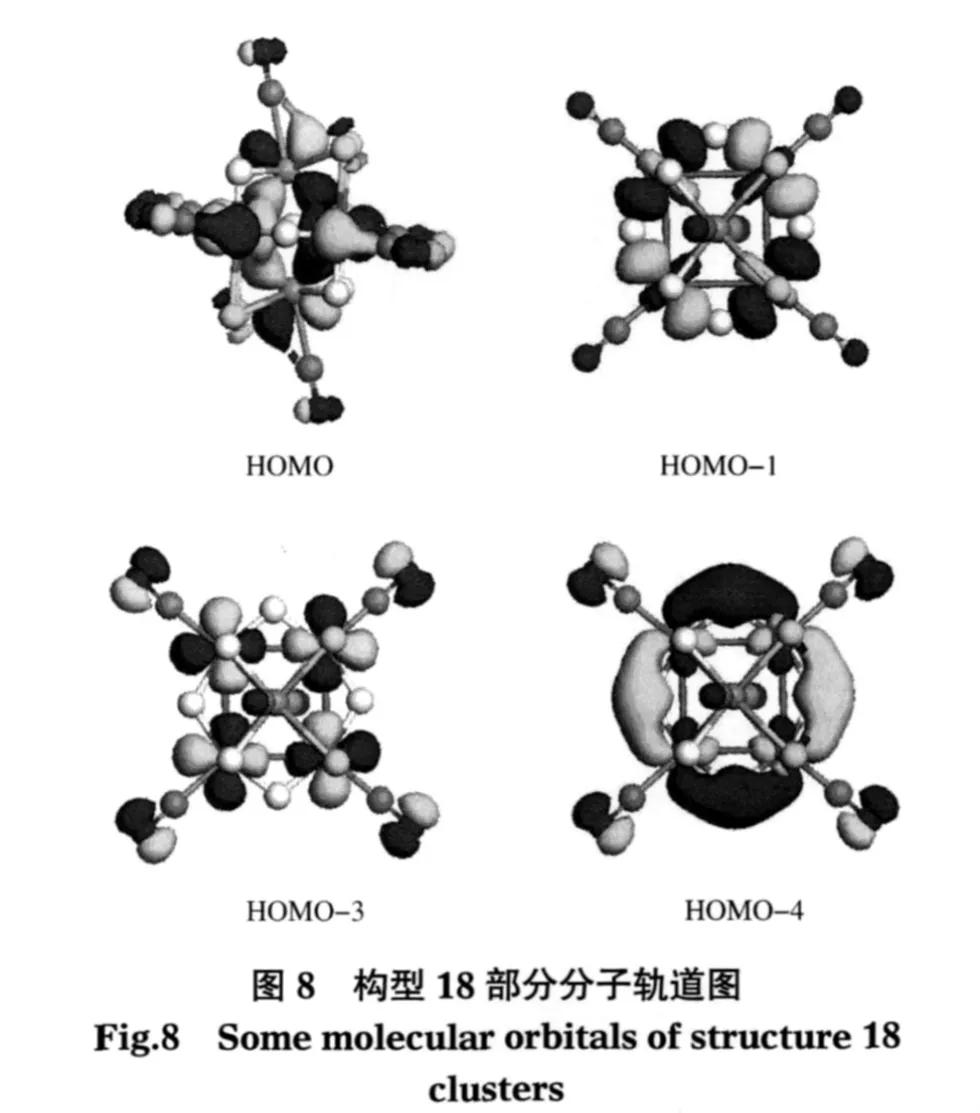
图8列出了构型18中的部分前线轨道,由此可以看出:混合团簇的前线轨道均主要由骨架金属原子Ta、Rh的d电子贡献;Ta原子和Rh原子间电子云有较大重叠;Rh原子间电子云均不重叠.可以推断钽铑混合团簇中多出的骨架电子应排在外层骨架反键(或非键)轨道上,故混合钽铑团簇的正八面体构型不稳定,发生畸变.计算证实所有二元钽铑混合团簇稳定构型对称性均较低,除构型18为C2h群,19为C4v群外,其它构型多为C1群或Cs.另外,由于d电子数增多,前线轨道间能级相近,ΔEH-L均介于[Rh6H12(CN)6]4-(0.37 eV)和[Ta6Cl12(CN)6]4-(1.48 eV)之间.其中构型1的ΔEH-L最小,为0.52 eV.构型22的ΔEH-L最大,为1.00 eV.
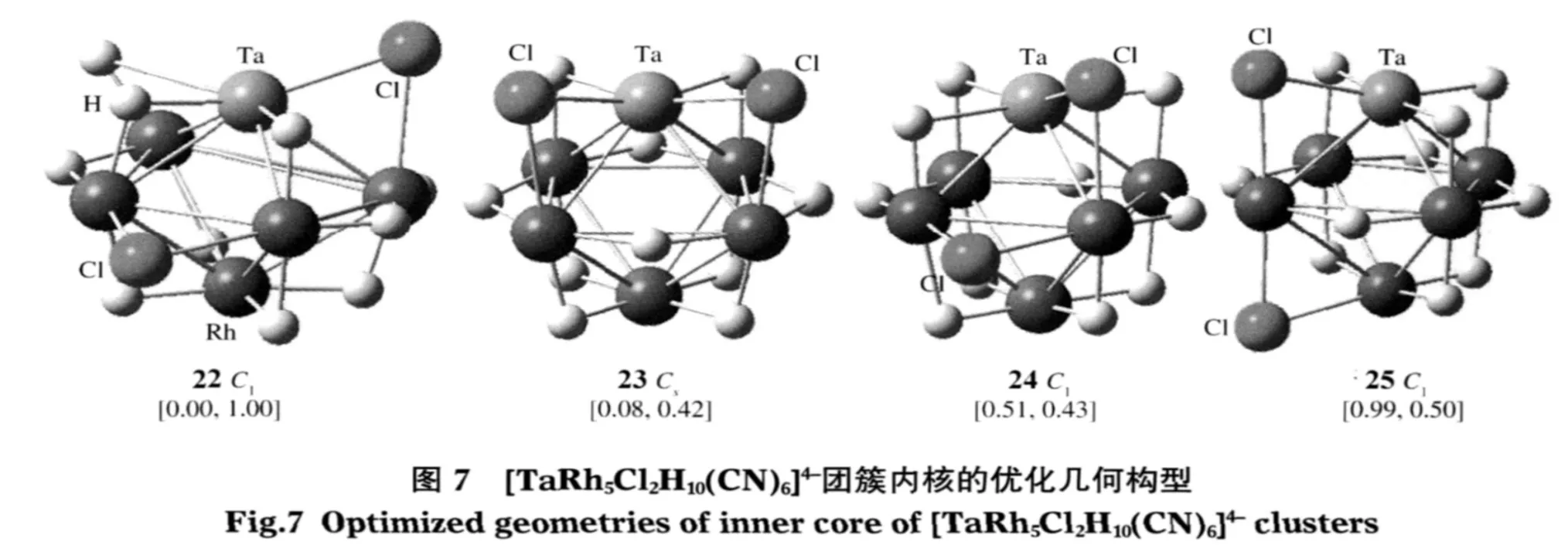
表3 钽铑簇合物的能隙(ΔEH-L)、平均Mulliken原子电荷(及平均Mulliken键级(MBO)Table 3 Energy gaps(ΔEH-L),average Mulliken atomic charges(),and average Mulliken bond orders(MBO)for Ta/Rh clusters
表3 钽铑簇合物的能隙(ΔEH-L)、平均Mulliken原子电荷(及平均Mulliken键级(MBO)Table 3 Energy gaps(ΔEH-L),average Mulliken atomic charges(),and average Mulliken bond orders(MBO)for Ta/Rh clusters
ΔEH-L/eV Q/e MBO Ta Rh Ta—Ta Ta—RhRh—Rh[Ta6Cl12(CN)6]4- 1.48 0.06 0.16[Ta5RhCl10H2(CN)6]4- 0.52 0.15-0.79 0.12 0.19[Ta4Rh2Cl8H4(CN)6]4- 0.68 0.19-0.85 0.12 0.16 -0.06[Ta3Rh3Cl6H6(CN)6]4- 0.61 0.28-0.88 0.10 0.17 0.11[Ta2Rh4Cl4H8(CN)6]4- 0.65 0.43-0.89 0.18 0.06[TaRh5C12H10(CN)6]4- 1.00 0.44-0.88 0.20 0.09[Rh6H12(CN)6]4- 0.37 -0.78 0.12
3 结 论
Ta、Rh是d电子互补的两种前后过渡金属,我们课题组曾研究(TiNi)n、(NbRh)n等混合簇得知:电子互补的前后过渡金属一维、二维团簇有明显的强弱交替化学键.而本论文研究的Ta、Rh团簇已形成三维结构,比较它们的成键情况可看出:对于纯金属团簇,前过渡金属团簇比后过渡金属团簇化合物稳定,而前后过渡金属混合簇化合物稳定性介于两者之间.混合簇的前线轨道主要由骨架金属原子的d电子贡献,这些轨道具有骨架反键(或非键)轨道性质,故HOMO与LUMO间的能隙均较小.随着Rh原子替代Ta原子个数的递增,对于混合簇稳定构型Ta—Ta键所起的作用逐渐减少,而Ta—Rh键所起的作用增加,Rh原子间为非键或反键性质.除[Ta2Rh4Cl4H8(CN)6]4-混合簇稳定构型对称性较高外(C2h),其余混合簇稳定构型对称性均较低(C1或Cs).
1 Braunstein,P.;Oro,L.A.;Raithby,P.R.Metal clusters in chemistry.Weinheim:Wiley-VCH,1999
2 Yan,B.B.;Zhou,H.J.;Lachgar,A.Inorg.Chem.,2003,42:8818
3 Zhou,H.J.;Day,C.S.;Lachgar,A.Chem.Mater.,2004,16:4870
4 Yan,Z.H.;Day,C.S.;Lachgar,A.Inorg.Chem.,2005,44:4499
5 Zhang,J.J.;Lachgar,A.J.Am.Chem.Soc.,2007,129:250
6 Zhou,H.J.;Strates,K.C.;Muñoz,M.A.;Little,K.J.;Pajerowski,D.M.;Meisel,M.W.;Talham,D.R.;Lachgar,A.Chem.Mater.,2007,19:2238
7 Zhou,H.J.;Lachgar,A.Crystal Growth&Design,2006,6:2384
8 Zhang,J.J.;Zhao,Y.;Gamboa,S.A.;Lachgar,A.Crystal Growth&Design,2008,8:172
9 Zhang,J.J.;Day,C.S.;Harvey,M.D.;Yee,G.T.;Lachgar,A.Crystal Growth&Design,2009,9:1020
10 Ingleson,M.J.;Mahon,M.F.;Raithby,P.R.;Weller,A.S.J.Am.Chem.Soc.,2004,126:4784
11 Brayshaw,S.K.;Ingleson,M.J.;Green,J.C.;Raithby,P.R;Kohn,G.K.;Mcindone,J.S.;Weller,A.S.Angew.Chem.Int.Edit.,2005,44:6875
12 Brayshaw,S.K.;Green,J.C.;Hazari,N.;Mcindoe,J.S.;Marken,F.;Raithby,P.R;Weller,A.S.Angew.Chem.Int.Edit.,2006,45:6005
13 Brayshaw,S.K.;Ingleson,M.J.;Green,J.C.;Mcindoe,J.S.;Raithby,P.R;Kohn,G.K.;Weller,A.S.J.Am.Chem.Soc.,2006,128:6247
14 Mingos,D.M.P.;Lin,Z.Y.Z.Phys.D,1989,12:53
15 Lin,Z.Y.;Williams,I.D.Polyhedron,1996,15:3277
16 Brayshaw,S.K.;Harrison,A.;Mcindoe,J.S.;Marken,F.;Raithby,P.R.;Warren,J.E.;Weller,A.S.J.Am.Chem.Soc.,2007,129:1793
17 Brayshaw,S.K.;Green,J.C.;Hazari,N.;Weller,A.S.Dalton Trans.,2007,18:1781
18 Douglas,T.M.;Brayshaw,S.K.;Raithby,P.R.;Weller,A.S.Inorg.Chem.,2008,47:778
19 Wang,Y.P.;Gu,Y.B.;Lin,M.H.;Zhang,Q.E.Chem.J.Chin.Univ.,2002,23:543 [王艺平,顾勇冰,林梦海,张乾二.高等学校化学学报,2002,23:543]
20 Wang,X.;Lin,M.H.;Zhang,Q.E.Acta Chim.Sin.,2004,62:1689 [王 娴,林梦海,张乾二.化学学报,2004,62:1689]
21 Qiu,W.W.;Lin,M.H.Acta Phys.-Chim.Sin.,2008,24:1573[邱玮玮,林梦海.物理化学学报,2008,24:1573]
22 (a)Delley,B.J.Chem.Phys.,1990,92:508(b)Delley,B.J.Chem.Phys.,2000,113:7756
23 Zhang,Q.E.Journal of Xiamen University,1981,20:226[张乾二.厦门大学学报,1981,20:226]
24 Basson,S.S.;Leipoldt,J.G.Transit.Met.Chem.,1982,7:207
Geometric and Electronic Structures of Hexanuclear Binary Ta/Rh Mixed Clusters
CAO Fei1,2,*TAN Kai2LIN Meng-Hai2
(1College of Chemistry and Chemical Engineering,Jiujiang University,Jiujiang 332005,Jiangxi Province,P.R.China;2Departmental of Chemistry,College of Chemistry and Chemical Engineering,Xiamen University,Xiamen 361005,Fujian Province,P.R.China)
O641
Received:June 13,2010;Revised:August 6,2010;Published on Web:September 17,2010.
*Corresponding author.Email:jjyz2001@163.com;Tel:+86-792-8312863.
The project was supported by the National Natural Science Foundation of China(20873107,20373053).
国家自然科学基金(20873107,20373053)资助项目
