一氧化氮在Cu3Pt(111)表面的吸附行为
2010-10-14解秀娟陈文凯孙宝珍章永凡
解秀娟 陈文凯,* 孙宝珍 郭 欣 章永凡
(1福州大学化学系,福州 350108; 2华中科技大学煤燃烧国家重点实验室,武汉 410074)
一氧化氮在Cu3Pt(111)表面的吸附行为
解秀娟1陈文凯1,*孙宝珍1郭 欣2章永凡1
(1福州大学化学系,福州 350108;2华中科技大学煤燃烧国家重点实验室,武汉 410074)
运用密度泛函理论中广义梯度近似(GGA)的PW91方法,结合周期性平板模型,探讨了NO分子在Cu3Pt(111)表面上不同吸附位的吸附行为.结果表明:NO分子以N端朝下方式吸附在top-Pt以及hcp1和fcc2位(分别为表面Cu2Pt和Cu3簇)的吸附模式最稳定,吸附能分别为101.8、124.5和118.1 kJ·mol-1.对于hcp1和fcc2位的吸附,NO中的N原子分别与底物的Cu2Pt和Cu3簇成键.吸附前后的电荷布居、态密度和振动频率的分析结果表明,净电子从底物合金表面转移到NO,N—O键伸长,频率发生红移.合金Cu3Pt和纯贵金属Pt对NO的吸附性质相似.
密度泛函理论; 一氧化氮; 铂铜合金; 吸附
Abstract:The adsorption behavior of NO onto a Cu3Pt(111)surface was studied using a periodic slab model and the PW91 generalized gradient approximation(GGA)within the framework of density functional theory.The calculated results indicated that NO adsorption with N-down on the top-Pt,hcp1,and fcc2 sites resulted in favorable structures with predicted adsorption energies of 101.8,124.5,and 118.1 kJ·mol-1,respectively.For adsorption onto the hcp1 and fcc2 sites,N atom from NO formed bonds with Cu2Pt and Cu3clusters,respectively.An analysis of the density of states,charge population,and vibrational frequencies before and after the adsorption showed that electrons transferred from the surface of the alloy to NO and that the N—O bond was elongated and its vibrational frequency was redshifted.The Cu3Pt alloy and pure precious metal Pt have similar adsorption properties to NO.
Key Words: Density functional theory;NO;Cu3Pt alloy; Adsorption
工业领域中,金属的合金化作为一种有效提高表面反应活性的途径,几十年来得到广泛的关注和研究[1-3].相对于纯金属,合金表面的几何和电子结构发生变化,从而改变合金的选择和催化性能.一直以来,Pt-Cu体系作为典型的二元合金体系得到广泛研究[4-6].通过探索Pt-Cu合金体系不同组分之间的电子相互作用,表明Pt-Cu合金体系作为烃重整催化剂,对石脑油脱氢裂解、CO的氧化、NOx的储存还原以及氢促进的氯代烃的脱氯反应起到催化作用[7].Shen等[8]通过低能离子散射(LEIS)实验研究发现Pt-Cu合金体系在高温条件下,形成连续取代的无规则结构.但是,在低温条件下三种有序相形成,分别为Cu3Pt,CuPt和Pt3Cu.此后,Cu3Pt体系引起研究者的极大兴趣.Shen等[9]对CO在Cu3Pt(111)表面的吸附行为进行LEIS实验研究.近年来Zhang等[10]通过密度泛函理论(DFT)研究CO分子在Cu3Pt(111)合金表面的氧化并与纯金属Pt(111)和Cu(111)表面进行比较.研究表明,CO在Cu3Pt(111)合金表面的吸附能低于在两种纯金属表面的吸附能.同时,Cu3Pt(111)合金表面上CO氧化的势垒低于纯金属表面,推断Cu3Pt合金是一种比Pt和Cu更好的催化剂.Linke[11]和Dino[12]等分别从实验和理论方面研究H2在Cu3Pt(111)合金表面吸附和解离,结果表明Cu3Pt(111)对H2的解离有很好的催化作用.Basile等[13]运用X射线衍射(XRD)等实验方法研究Pt-Cu/水滑石对NOx的储存还原的催化作用,研究表明Pt-Cu合金的形成不仅使其催化性能较好,且可以防止催化剂的钝化.金属Pt对NOx的储存还原具有很高的催化活性[14-19],而且金属Cu是一种廉价金属,因此,Pt-Cu合金对NOx的储存还原催化性质的研究显得十分重要.因为大气中NOx(其中NO占90%以上)的浓度随着近年来燃煤烟气和汽车尾气的过度排放而呈现明显的上升趋势,且NO在太阳光作用下,极易形成光化学烟雾.目前对NO分子在Cu3Pt合金表面上的吸附分解的理论研究还未见报道.
本文采用密度泛函理论结合周期平板模型方法,模拟NO分子在Cu3Pt(111)表面的吸附行为和解离趋势,对NO分子在Cu3Pt(111)表面不同吸附位上的吸附构型进行了构型优化以及电荷布居、态密度、轨道成分和振动频率的计算,并讨论了不同吸附位的稳定性和吸附能,初步探讨NO分子与Cu3Pt(111)表面相互作用的实质.
1 计算模型和方法
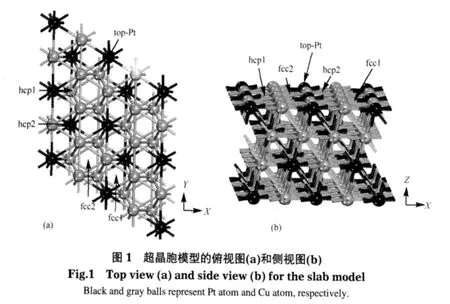
Cu3Pt类似Cu3Au型,属于面心立方堆积,Pt原子置于顶角位,Cu原子置于晶胞的面心位[20].在整齐有序的Cu3Pt(111)表面,Pt原子周围均为Cu原子[21-22].本文选取如图1所示的Cu3Pt(111)2×2超晶胞四层平板模型研究NO分子在Cu3Pt(111)表面的吸附.
本文采用广义梯度密度泛函理论和周期平板模型方法模拟NO分子在Cu3Pt(111)表面的吸附.所有的计算均由Dmol3软件包[23-24]实现.计算采用PW91泛函,Pt和Cu原子内层电子由有效核电势(ECP)代替,价电子波函数采用双数值基加极化函数(DNP)展开,非金属原子N和O采用全电子基组.Brillouin zone积分的Monkhorst-Pack网格参数设为 3×3×1,Methfessel-Paxton smearing 设为 0.005 Ha;结构优化以能量、位移和力收敛为判据,收敛标准分别为 1×10-5Ha,5×10-4nm,2×10-4Ha·nm-1,结构优化时最底下三层原子固定在体相的位置,上面一层及N和O原子的坐标全部放开.相邻两层平板间的真空度厚度为1.3 nm,以确保平板间相互作用足够小.
吸附能定义为吸附前后各物质总能量的变化:
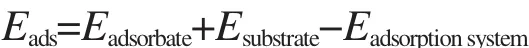
其中Eadsorbate和Esubstrate分别表示吸附前吸附质和底物的能量,Eadsorptionsystem表示吸附后体系的总能量.
2 结果与讨论
2.1 NO分子在Cu3Pt(111)表面的吸附构型和吸附能
考虑NO在如图1所示Cu3Pt(111)表面的五种不同吸附位,分别是top-Pt,fcc1,fcc2,hcp1,hcp2,其中1代表1个Pt原子和2个Cu原子构成的穴位,2代表3个Cu原子构成的穴位.在每个吸附位上,考虑NO分子三种初始吸附取向:N端朝下,O端朝下和NO平行底物表面.构型优化后,NO分子以N端朝下的吸附是化学吸附,O端朝下为物理吸附,平行方式均转变为N端朝下.因此,为简明起见,在0.25 Pt单层覆盖度下(0.25 ML),所考察的三种化学稳定吸附构型的构型参数和吸附能如图2所示.
由图2可见,吸附能的大小顺序为hcp1>fcc2>top-Pt,吸附能愈大,吸附分子与衬底之间的吸附愈强,由此可见NO以N端在hcp1位的吸附最强.值得指出的是,NO在hcp1位和fcc2位及其top-Pt位的吸附能差值不是非常大,因此NO以N端吸附在这三种位置的吸附模式在能量上比较有利,均为稳定的吸附构型.
对于N—O键,NO在Cu3Pt(111)表面的这3个吸附位的吸附均使N—O键相对于自由NO分子(计算值为0.1163 nm,实验值为0.1151 nm[25])伸长,但top-Pt位上N—O键长变化并不明显,而在hcp1和fcc2位N—O键长变化非常显著,而且N—O键长均在(0.118±0.004)nm范围,这与实验测得NO在纯金属Pt(111)的吸附键长范围[26]相吻合.可见NO在Cu3Pt(111)表面的吸附和在Pt(111)表面的吸附同样造成了NO的N—O键削弱,有利于NO的进一步解离.文献[10]中,CO稳定吸附在Cu3Pt(111)表面top-Pt位的吸附能为1.51 eV(145.7 kJ·mol-1),其大于本文NO在Cu3Pt(111)表面top-Pt位的吸附能(101.8 kJ·mol-1),但根据N—O和C—O键的活化难易程度,N—O键可能更容易,这是因为NO分子的解离能远低于CO分子的解离能.
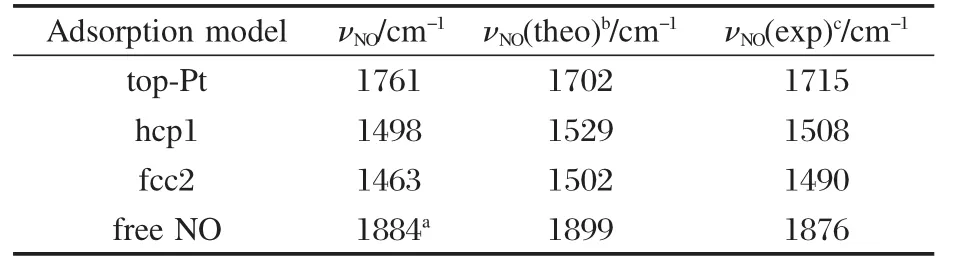
表1 NO分子在Cu3Pt(111)和Pt(111)不同吸附位的振动频率Table 1 Vibrational frequencies of NO on different adsorptional sites for Cu3Pt(111)and Pt(111)surfaces
2.2 NO的伸缩振动频率分析
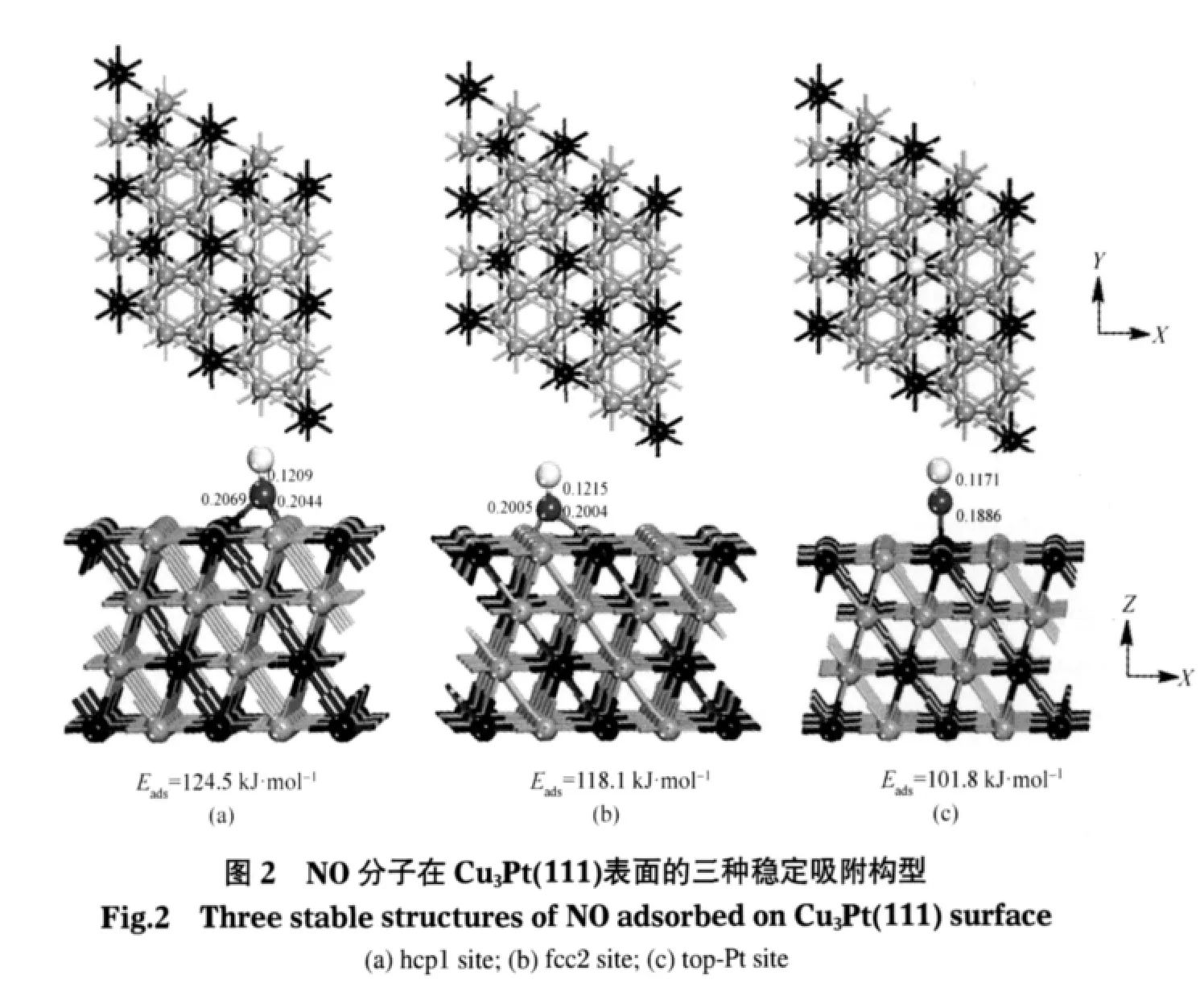
表1为NO在Cu3Pt(111)表面不同吸附位的振动频率和NO在Pt(111)表面的理论[18]及实验[27]振动频率值.由表可见,我们计算得出自由分子νNO为1884 cm-1,与实验值1876 cm-1相符合.对于Cu3Pt(111)表面,NO吸附后,频率红移,尤其在hcp1和fcc2稳定吸附位,红移了400 cm-1左右,这与N—O键长伸长,键被削弱的结果相符合.而且NO频率红移的大小与键长的伸长程度及吸附能大小的变化趋势一致.Getman等[18]用DFT研究NO在Pt(111)表面的吸附,计算得出的top,hcp和fcc吸附位NO的振动频率的变化趋势与我们的结果一致,并且均与实验值一致.以上事实进一步说明Cu3Pt(111)表面对NO的吸附与纯贵金属Pt(111)表面的吸附性质相近.从而我们推测Cu3Pt合金和纯贵金属Pt对NO的催化性能相似.

表2 NO中各原子和Cu及Pt原子的平均Mulliken电荷布居Table 2 Average Mulliken populations for N,O in NO and Cu,P atoms
2.3 Mulliken电荷布居和轨道布居分析
表2给出了吸附前后NO各原子和底物金属原子的Mulliken电荷布居,由表2可见,吸附后,NO既是得电子体,又是失电子体.因为NO分子的电子组态为:NO[(1σ)2(2σ)2(3σ)2(4σ)2(1π)4(5σ)2(2π)1][28],即最高占据轨道(HOMO)和最低未占据轨道(LUMO)均为半充满的2π反键轨道.这意味着当NO分子吸附在固体表面时,它既可以从表面接受电子,又可以将电子反馈到固体表面.在hcp1和fcc2两个吸附位,NO均得到电子,Pt和Cu原子均失去电子.值得注意的是,在top-Pt吸附位,NO分子和Pt原子均失去电子,为了解释这个看似矛盾的结论,表3给出NO吸附前后的轨道布居.由表可知,与自由NO分子相比,处于top-Pt位吸附时N原子和O原子的2px,2py轨道均得到电子,根据我们实验组之前的研究[29]表明,NO分子的2π反键轨道是由N和O原子的2p轨道杂化形成.由此可以得出NO分子在top-Pt位吸附后其2π反键轨道得到电子,导致键级变小,N—O键长伸长.但是在Getman等[18]的文献报道中,NO在Pt(111)表面的top,hcp及fcc位的吸附,均为得到电子.说明Cu和Pt合金化在一定程度上改变了NO电子的转移方式.
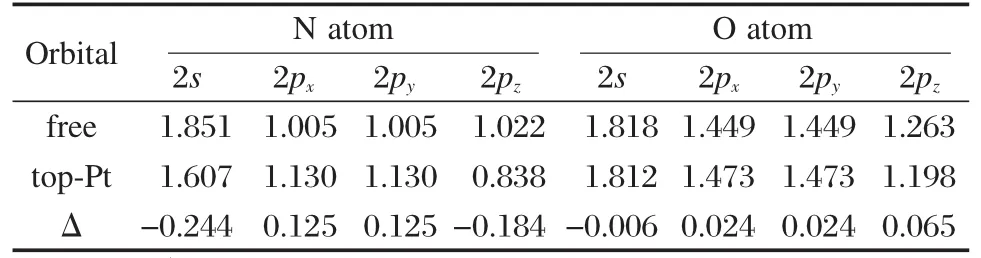
表3 N和O原子在top-Pt位吸附前后的轨道布居Table 3 Orbital populations for N and O atoms on top-Pt site before and after adsorption

2.4 态密度分析
图3给出了吸附前后NO分子和吸附位Pt及Cu原子的态密度图.与吸附前的态密度相比,吸附后的的NO分子的2π反键轨道的态密度峰发生了明显的离域化,与Cu 3d和Pt 5d轨道的态密度峰发生明显重叠,说明NO分子在吸附后与Cu及Pt原子形成化学键,且造成N—O键长伸长,频率红移,这与我们前面分析的相符合.NO分子发生红移是由于电子流从Cu3Pt表面转移到吸附分子所导致,即NO分子得到电子,这与上述Mulliken电荷分析结果相一致.在文献[10]中,通过对比纯金属Pt和合金Cu3Pt中Pt原子的态密度,表明合金化之后,Pt原子的带中心向低能级移动了0.12 eV.这就稍微降低了Pt的d轨道与NO的2π反键轨道的成键作用,即NO在合金Cu3Pt上的吸附会稍弱于在纯金属Pt上的吸附.
3 结 论
采用密度泛函理论,模拟了NO分子吸附在Cu3Pt(111)表面的平衡几何结构和相应的电子结构.对NO分子在Cu3Pt(111)表面不同吸附位上的吸附进行了构型优化.结果表明,NO分子以N端朝下方式吸附在top-Pt,hcp1和fcc2位最稳定,且N—O键伸长,有进一步解离的可能.Mulliken电荷布居分析结果表明,吸附过程中NO既是得电子体又是失电子体.振动频率的结果表明,吸附后NO频率发生红移,进一步说明N—O键的削弱.并且通过与文献报道中NO在Pt(111)表面吸附的比较,表明Cu3Pt合金和纯贵金属Pt对NO的吸附性质相似,进一步推测它们对NO的催化性能也可能相似.当然,这还取决于NO后面的解离、成键和脱附行为.
1 Ponec,V.Catal.Rev.,1975,11:41
2 Campbell,C.Annu.Rev.Phys.Chem.,1990,41:775
3 Bardi,U.Rep.Prog.Phys.,1994,57:939
4 Knudsen,J.;Nilekar,A.U.;Vang,R.T.;Schnadt,J.;Kunkes,E.L.;Dumesic,J.A.;Mavrikakis,M.;Besenbacher,F.J.Am.Chem.Soc.,2007,129:6485
5 Luyten,J.;Schurmans,M.;Creemers,C.;Bunnik,B.S.;Kramer,G.J.Surf.Sci.,2007,601:2952
6 Pasti,I.;Mentus,S.Mater.Chem.Phys.,2009,116:94
7 Avdeev,V.I.;Kovalchuk,V.I.;Zhidomirov,G.M.;d′Itri,J.L.Surf.Sci.,2005,583:46
8 Shen,Y.G.;O′Connor,D.J.;Wandelt,K.;MacDonald,R.J.Surf.Sci.,1995,328:21
9 Shen,Y.G.;O′Connor,D.J.;Wandelt,K.;MacDonald,R.J.Surf.Sci.,1995,331-333:746
10 Zhang,C.J.;Baxter,R.J.;Hu,P.;Alavi,A.;Lee,M.H.J.Chem.Phys.,2001,115:5272
11 Linke,R.;Schneider,U.;Busse,H.;Becker,C.;Schröder,U.;Castro,G.R.;Wandelt,K.Surf.Sci.,1994,307-309:407
12 Dino,W.A.;Kasai,H.;Okiji,A.Surf.Sci.,2001,482-485:318
13 Basile,F.;Fornasari,G.;Livi,M.;Tinti,F.;Trifiro,F.;Vaccari,A.Top.Catal.,2004,30/31:223
14 Abe,A.;Yamashita,K.Chem.Phys.Lett.,2004,393:331
15 Zhu,P.;Shimada,T.;Kondoh,H.;Nakai,I.;Nagasaka,M.;Ohta,T.Surf.Sci.,2004,565:232
16 Jiang,Z.;Huang,W.;Tan,D.;Zhai,R.;Bao,X.Surf.Sci.,2006,600:4860
17 Tsukahara,N.;Mukai,K.;Yamashita,Y.;Yoshinobu,J.;Aizawa,H.Surf.Sci.,2006,600:3477
18 Getman,R.B.;Schneider,W.F.J.Phys.Chem.C,2007,111:389
19 Smeltz,A.D.;Getman,R.B.;Schneider,W.F.;Ribeiro,F.H.Catal.Today,2008,136:84
20 Hansen,M.;Anderko,K.Constitution of binary alloys.2nd ed.New York:McGraw-Hill,1958
21 Castro,G.R.;Schneider,U.;Busse,H.;Janssens,T.;Wandelt,K.Surf.Sci.,1992,269-270:321
22 Schneider,U.;Castro,G.R.;Busse,H.;Janssens,T.;Wesemann,J.;Wandelt,K.Surf.Sci.,1992,269-270:316
23 Delley,B.J.Chem.Phys.,1990,92:508
24 Delley,B.J.Chem.Phys.,2000,113:7756
25 Lide,D.CRC handbook of chemistry and physics.Boca Raton:CRC Press,2003:9-20
26 Materer,N.;Barbieri,A.;Gardin,D.;Starke,U.;Batteas,J.D.;Vanhove,M.A.;Somorjai,G.A.Physical Review B,1993,48:2859
27 Matsumoto,M.;Tatsumi,N.;Fukutani,K.;Okano,T.Surf.Sci.,2002,513:485
28 Pan,D.A.;Zhao,C.D.;Zheng,Z.X.Structures of matters.2nd ed.Beijing:Higher Education Press,1989:200-201 [潘道皑,赵成大,郑载兴.物质结构.北京:高等教育出版社,1989:200-201]
29 Sun,B.Z.;Chen,W.K.;Zheng,J.D.;Lu,C.H.Appl.Surf.Sci.,2008,255:3141
Adsorption Behavior of NO on Cu3Pt(111)Surface
XIE Xiu-Juan1CHEN Wen-Kai1,*SUN Bao-Zhen1GUO Xin2ZHANG Yong-Fan1
(1Department of Chemistry,Fuzhou University,Fuzhou 350108,P.R.China;2State Key Laboratory of Coal Combustion,Huazhong University of Science and Technology,Wuhan 410074,P.R.China)
O641
Received:July 12,2010;Revised:August 28,2010;Published on Web:September 30,2010.
*Corresponding author.Email:qc2008@fzu.edu.cn;Tel:+86-591-22866162.
The project was supported by the National Natural Science Foundation of China(90922022),State Key Laboratory of Coal Combustion Foundation of Huazhong University of Science and Technology,China(FSKLCC0814),and New Century Excellent Talents Program in University of Fujian
Province,China(HX2006-103,HX2006-98).
国家自然科学基金(90922022),华中科技大学煤燃烧国家重点实验室基金(FSKLCC0814)和福建省高等学校新世纪优秀人才计划(HX2006-103,HX2006-98)资助项目
