第4 代小分子表皮生长因子受体-酪氨酸激酶抑制剂研究进展
2023-08-10邓连力
邓连力
(重庆市秀山土家族苗族自治县人民医院,重庆 409900)
癌症是全世界人类死亡的主要原因之一,全球癌症的发病率和死亡率均呈上升趋势[1-2]。表皮生长因子受体(EGFR)在癌细胞的增殖、存活、黏附、迁移和分化过程中起重要作用,是治疗癌症的有效靶点[3-4],以EGFR 为靶点进行药物研发是抗肿瘤药物研究的热点。第1,2,3 代表皮生长因子受体- 酪氨酸激酶抑制剂(EGFR-TKIs)已被批准用于癌症的治疗,代表药物有吉非替尼、阿法替尼和奥希替尼等,显著提高了癌症患者的客观缓解率,延长了患者的无进展生存期,治疗效果良好[5],但易产生获得性耐药[6]。为此,研发第4 代小分子EGFR-TKIs 是目前急需解决的问题。本研究中检索了PubMed,SCI - hub,CNKI 数据库中自建库起至2022 年9 月第4 代小分子EGFR - TKIs 的相关研究文献,总结其研究现状。现报道如下。
1 EGFR 概述与常见突变类型
1962 年,Stailley Cohen 发现了EGFR,并确认其为生长因子[7]。EGFR 是一种跨膜蛋白,将生长因子信号从细胞外部传入到细胞内部,与肿瘤的生长与转移密切相关。EGFR 在癌细胞膜表面高度表达,且在肿瘤细胞的生长与分化中起重要作用,是肿瘤诊断和治疗的靶点[8]。EGFR 位于细胞膜表面,通过与配体结合来激活。激活后,EGFR 由单体转化为二聚体,并导致酪氨酸残基磷酸化,活化受体的磷酸酪氨酸残基又是靶分子的对接位点。靶分子与靶标的结合导致RAS/RAF 和磷脂酰肌醇3 激酶- 丝氨酸/ 苏氨酸蛋白激酶(PI3K -AKT)信号级联通路激活,调节癌细胞的增殖、存活、黏附、转移与分化[9]。EGFR - TKIs 通过与细胞内酪氨酸激酶结构域上三磷酸腺苷(ATP)位点竞争性结合,抑制受体自身磷酸化,阻断酪氨酸激酶的活化,从而抑制肿瘤细胞周期的进程,加速肿瘤细胞凋亡[10]。
EGFR基因位于7号染色体短臂上,其中外显子18-21区在治疗过程中常伴有突变。生物标志物和突变分析法研究表明,外显子19 区的缺失突变、外显子18 区和21 区的单核苷酸的替换突变及T790M 突变,占据了突变类型的90%[11-14];外显子21 区的L858R 位点突变的发生率为40%[15]。T790M 突变是第1,2 代EGFR-TKIs产生耐药最常见的机制,C797S 突变是第3 代EGFR -TKIs 产生耐药的主要机制[16-19]。上述耐药现象的产生,促进了新一代EGFR-TKIs的诞生。
2 第4 代小分子EGFR-TKIs 研究进展
2.1 整体研究进展
目前,应用于新型C797S 突变的第4 代EGFR -TKIs 尚未上市,但关于克服此种突变的研究已取得一定进展。大多数第4 代小分子EGFR-TKIs 通过计算机辅助药物设计得到,其作用机制为药物小分子可与ATP竞争性结合于EGFR 的细胞内催化结构域,从而抑制EGFR的磷酸化和下游信号传递[20],从信号通路上游阻断下游级联反应,最终抑制癌细胞的分化与增殖。详见图1。
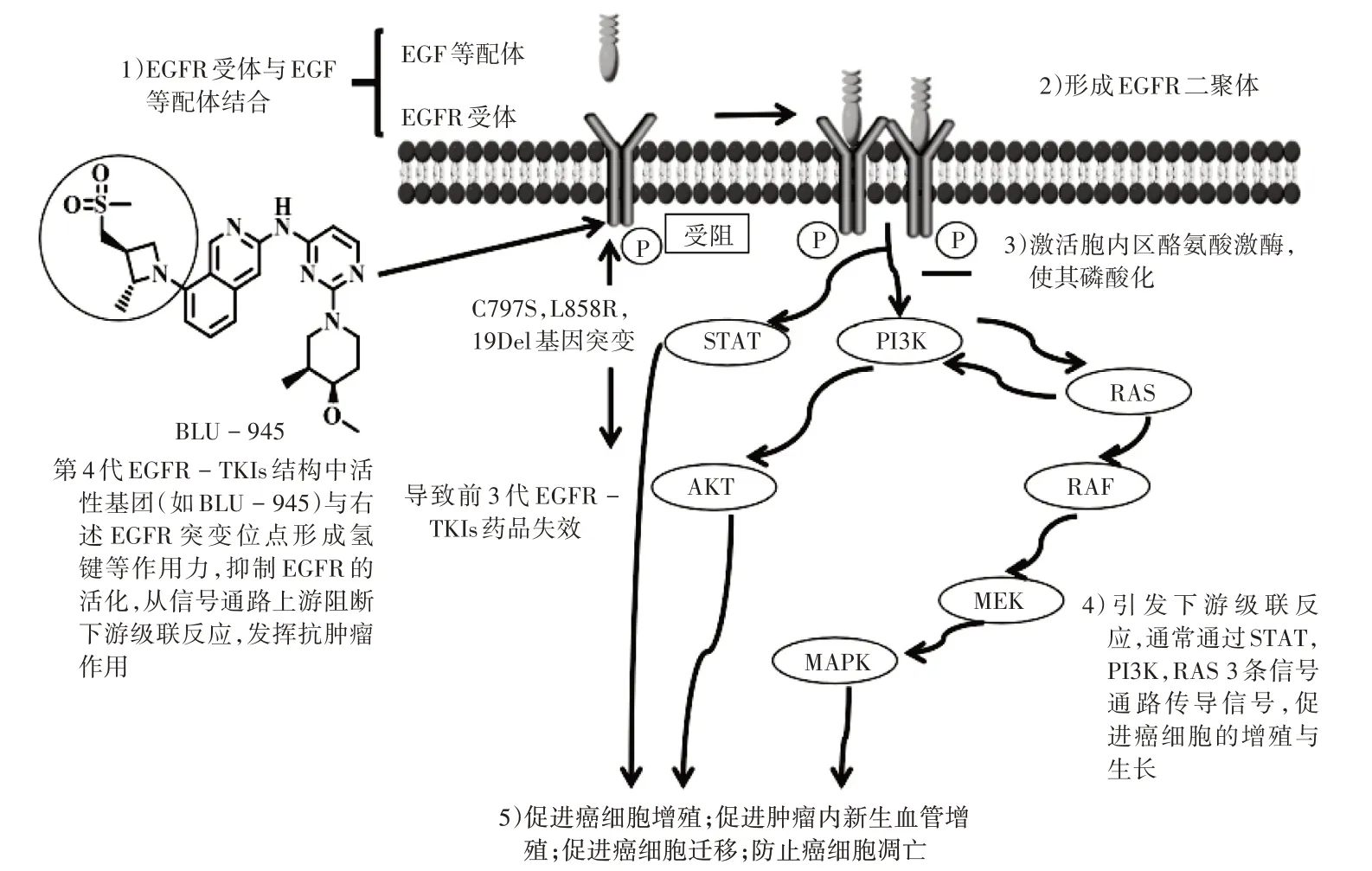
图1 第4代小分子EGFR-TKIs的作用机制Fig.1 Mechanism of the fourth generation of small molecule EGFR-TKIs
2.2 临床研究的第4 代小分子EGFR-TKIs
2.2.1 ES-072
ES - 072 是由浙江博生医药有限公司自主研发的第4 代EGFR - TKIs,是一种不可逆的突变体选择性EGFR 共价抑制剂,可选择性抑制突变的EGFR L858R,19Del和EGFR T790M,抑制T790M/L858R/C797S突变的NCI-H1975细胞的半数抑制度(IC50)为1.06µmol/L。2020 年,ZHENG 等[21]在第1,2 代EGFR 抑制剂治疗失败的T790M 突变的非小细胞肺癌(NSCLC)患者中首次完成人体Ⅰ期临床试验(CTR20180074)。研究结果表明,ES-072 在NSCLC 患者中安全且耐受性良好,不良事件多为1 级或2 级,后续治疗后可逆转;与第1 代EGFR-TKIs 相比,ES-072 治疗后皮疹和腹泻的发生率分别为15.8%和10.5%,而吉非替尼上述2种不良反应的发生率均为50%,提示ES-072对突变的EGFR选择性更高。值得注意的是,ES-072 具有与第3 代EGFRTKIs 相似的心脏毒性,表现为高发生率(57.9%)的QT间期延长,在后续临床试验中需加强对患者的心脏功能指标的监测。药代动力学方面,ES - 072 的半衰期为24.5 h,适合每日给药1次,达峰时间约为4 h;药效动力学方面,ES - 072 对NSCLC 患者的缓解率和控制率分别为46.2%和76.9%,而奥希替尼分别为62%和90%,抗肿瘤效果低于奥希替尼。化学结构见图2 A。

A.ES-072 B.TQB3804 C.BLU-945图2 开展临床研究的第4代小分子EGFR-TKIs的化学结构A.ES-072 B.TQB3804 C.BLU-945Fig.2 Chemical structures of the fourth generation of small molecule EGFR -TKIs that have been studied in clinical studies
2.2.2 TQB3804
TQB3804 是由正大天晴药业集团股份有限公司与药明康德新药开发研究公司联合研发的第4 代EGFRTKIs,在2019年的美国癌症研究协会(AACR)年会上首次报道。其不仅能克服导致第3 代EGFR-TKIs 耐药的2 种常见三重突变(19Del/ T790M/ C797S 和L858R/T790M/ C797S,IC50< 0.1 nmol/ L),且 抑 制EGFR T790M 突变的效果较其他EGFR-TKIs 更佳。提示该药对于第1,2,3 代EGFR - TKIs 耐药都有不错的抑制能力。在EGFR Del746-750/T790M/C797S 突变的Ba/F3细胞系异种移植模型实验中,免疫印迹法分析结果显示,TQB3804 通过抑制磷酸化EGFR(p - EGFR)、磷酸化AKT(p - AKT)、磷酸化细胞外信号调节激酶(p -ERK)靶向三联抑制突变的EGFR,可较好地抑制移植肿瘤模型生长,而奥希替尼无法阻止C797S突变肿瘤的生长[22]。2019 年11 月,开始TQB3804 的Ⅰ期临床试验(NCT04128085),目前尚无安全性及对C797S 突变耐药癌症患者的缓解率和控制率的研究报道。化学结构见图2 B。
2.2.3 BLU-945
BLU - 945 是Blueprint Medicines 公司研发 的 专 门针对奥希替尼耐药后继发的T790M/ C797S 共发突变及其他T790M 耐药突变的第4 代EGFR 抑制剂。ENO等[23]研究发现,BLU-945 抑制L858R/T790M/C797S和19Del/ T790M/ C797S 突变的Ba/ F3 细胞系的IC50分别为3.20 nmol/L 和4.00 nmol/L。在Ba/F3 L858R/T790M/C797S 和Ba/F3 19Del/T790M/C797S突变的奥希替尼耐药肿瘤模型小鼠中,BLU-945(100 mg/kg,每日2 次)产生了强烈的肿瘤消退作用,而奥希替尼未显示出任何抗肿瘤作用。在奥希替尼耐药的NSCLC患者细胞来源的异种移植模型中,用BLU - 945(75,100 mg/kg,每日2 次)治疗模型小鼠56 d,观察到抑制肿瘤生长的作用。
2021 年6 月,开始该药的Ⅰ期/ Ⅱ期临床试验(NCT04862780)。研究结果表明,BLU-945(25~400 mg,每日1 次)在奥希替尼耐药的NSCLC 患者中安全且耐受性良好,未发现4 级和5 级不良反应,也未发现QT 间期延长不良事件,皮疹和腹泻的发生率均不超过10%;高剂量(200~400 mg,每日1 次)BLU-945 治疗患者的肿瘤明显缩小,抗肿瘤活性强。通过测序分析患者血浆中的ctDNA 发现,BLU - 945 治疗后14 d,患者血样中T790M 和C797S 突变丰度下降,表明BLU - 945 对于T790M 和C797S突变的患者具有抑制作用。相比其他在研的第4 代EGFR -TKIs,BLU - 945 研究进展最快,安全性、有效性较好。化学结构见图2 C。
2.3 临床前研究的第4 代小分子EGFR-TKIs
2.3.1 EAI045
JIA 等[24]用纯化的EGFR L858R/T790M 激酶筛选出的化合物中,鉴定出1 种新的EGFR 变构抑制剂- 1(EAI001),且对EGFR L858R/ T790M 突变有活性。进一步优化得到变构抑制剂EAI045[25],该抑制剂抑制EGFR L858R/ T790M 突变的IC50为3.00 nmol/ L。在L858R/T790M 突变的肺癌模型小鼠中,EAI045 与西妥昔单抗联合治疗L858R/T790M 突变的模型小鼠,观察到肿瘤明显消退,而单用EAI045 未见肿瘤消退,且在L858R/ T790M/ C797S 突变的肺癌模型小鼠中观察到相似的效果。上述结果表明,EAI045 可克服T790M 和C797S 突变的耐药性,必须联用西妥昔单抗才有效[24,26],但联合用药的不良反应较大,故EAI045 的临床试验未能成功开展[27]。化学结构见图3 A。

A.EAI045 B.JBJ-04-125-02 C.BI-4020 D.JND3229 E.CH7233163 F.AZ7608图3 开展临床前研究的第4代小分子EGFR-TKIs的化学结构A.EAI045 B.JBJ-04-125-02 C.BI-4020 D.JND3229 E.CH7233163 F.AZ7608Fig.3 Chemical structures of the fourth generation of small molecule EGFR -TKIs in preclinical studies
2.3.2 JBJ-04-125-02
TO 等[28]在变构抑制剂EAI045 的基础上优化改造,得到了变构抑制剂JBJ - 04 - 125 - 02。用L858R,L858R/ T790M,L858R/ T790M/ C797S 突变稳定转染的Ba/F3 细胞系测试结果表明,与EAI045 联用或不联用西妥昔单抗相比,JBJ - 04 - 125 - 02 是可以作为单一药物抑制细胞增殖的化合物,且抑制上述3种突变的IC50分 别 为1.00,0.26,0~0.10 nmol/ L。在L858R/T790M/C797S 突变基因工程小鼠中的研究结果表明,50 mg/kg JBJ-04-125-02 可较好抑制肿瘤生长,提示JBJ - 04 - 125 - 02 对C797S 突变具有抑制能力。另外,基于JBJ-04-125-02及奥希替尼的结构,两药联用能发挥协同增效作用,奥希替尼能促使JBJ - 04 -125-02更多、更好地与癌细胞结合,起到增敏效果,且两药联用对癌细胞生长的抑制能力更强,能更有效地缩小肿瘤体积。提示共价突变选择性ATP 竞争抑制剂(如奥希替尼)联用变构抑制剂可能是EGFR 突变癌症患者的有效治疗方法,后续仍需进行大量的临床研究。化学结构见图3 B。
2.3.3 BI-4020
ENGELHARDT 等[29]从选择性激酶抑制剂库中筛选优化出一种高选择性苯并咪唑化合物BI - 4020。BI - 4020 是一种非共价大环TKIs,抑制EGFR 19Del/T790M/C797S突变的Ba/F3细胞的IC50为0.20 nmol/L。EGFR 19Del/T790M/C797S突变的PC-9 细胞异种移植肿瘤模型小鼠中,BI-4020 组(10 mg/kg,每日1 次,灌胃)小鼠治疗19 d 后的肿瘤明显消退[肿瘤抑制率(TGI)为121%]。结果表明,BI - 4020 诱导肿瘤消退明显,且耐受性良好,而奥希替尼组(25 mg/kg,每日1次,灌胃)模型小鼠的肿瘤消退作用并不明显。提示BI-4020对T790M 和C797S 突变均有抑制作用,可能会在奥希替尼耐药且目前没有TKIs 靶向治疗选择的临床环境中发挥作用。化学结构见图3 C。
2.3.4 JND3229
JND3229 是一种可逆的EGFR 突变抑制剂,对L858R/ T790M/ C797S 突变的IC50为5.80 nmol/ L。在小鼠实验中,JND3229可明显抑制BaF3 19Del/T790M/C797S 异种移植BALB/ c 小鼠肿瘤的生长(IGF =42.2%),效果优于EAI045 联用西妥昔单抗(IGF =22.3%)。此外,JND3229 耐受性良好,在小鼠中没有明显的体质量减轻或其他毒性迹象。通过X 射线发现,JND3229 被容纳在具有可逆“U 形”构型的C797S 突变EGFR 的ATP 结合位点中,其吡啶[2,3 - d]嘧啶环与EGFR蛋白的第793位甲硫氨酸残基形成双氢键相互作用,吡啶[2,3-d]嘧啶环的羰基与EGFR 蛋白745 位赖氨酸残基的氮原子通过水分子介导形成氢键,丙酰胺基团中的氨基与EGFR 蛋白718位亮氨酸通过另1个水分子介导形成氢键,左侧甲基取代的苯基通过范德华力与796位甘氨酸相互作用[30]。该抑制剂为开发第4代C797S 突变的EGFR - TKIs 提供了重要的结构和化学基础。化学结构见图3 D。
2.3.5 CH7233163
KASHIMA[31]等设计合成了CH7233163,该化合物抑制19Del/T790M/C797S突变的IC50为0.28 nmol/L,呈剂量依赖性地阻断19Del/T790M/C797S NIH3T3 细胞中的EGFR及下游AKT和ERK1/2的磷酸化。在体内试验中,CH7233163[100 mg/(kg·d)]治疗19Del/T790M/C797S NIH3T3 异种移植的肿瘤模型小鼠,14 d 后肿瘤明显缩小。晶体结构分析结果显示,CH7233163 是一种非共价ATP 竞争的EGFR 19Del/ T790M/ C797S 抑制剂,利用与EGFR 的αC-螺旋构象的多重相互作用,有效抑制活性和突变选择性。化学结构见图3 E。
2.3.6 AZ7608
FLOCH 等[32]设计合成了化合物AZ7608,该化合物抑制PC9(19Del),PC9-TM(19Del/T790M/C797S),H1975(L858R/T790M),H1975-TM(L858R/T790M/C797S)4 种耐药细胞系的IC50分别为7.3,7.0,6.5,6.5 nmol/L,其抗肿瘤活性与布加替尼相似。AZ7608可显著抑制PC9-TM 和H1975细胞的异种移植模型的肿瘤生长,AZ7608联用西妥昔单抗可缩小82%的PC9-TM异种移植模型的肿瘤体积,成功克服C797S突变诱导的获得性耐药。该抑制剂可作为第4 代EGFR-TKIs 的研究方向。化学结构见图3 F。
3 结语
目前已上市的EGFR-TKIs 虽有治疗效果,但均会出现耐药性。第1,2代抑制剂耐药的主要机制为T790M突变,第3 代抑制剂耐药的主要机制为C797S 突变。以耐药机制为切入点,并将其作为相应位点可研制出新的治疗药物[24]。已报道的第4 代小分子EGFR - TKIs中,各抑制剂对肿瘤突变均有较好的抑制作用。已完成Ⅰ期临床试验的ES-072 和BLU-945 的安全性、有效性均较好,不良反应较轻,耐受性好。其余抑制剂尚未在人体中进行相关试验,但EAI045,JBJ-04-125-02等变构抑制剂联用共价突变选择性ATP 竞争抑制剂(如奥希替尼)起到了增敏作用,为第4 代EGFR-TKIs克服肿瘤耐药的研究提供了参考。未来的临床试验仍是各抑制剂被关注的重点,相信随着各种新EGFR -TKIs 临床试验的开展,EGFR 耐药难题将得到逐步解决。
