不同壁材对绿咖啡油微胶囊微观结构及其热稳定性的影响
2023-05-30慕静怡胡发广张珍珍董文江李亚男毕晓菲胡荣锁陈小爱
慕静怡 胡发广 张珍珍 董文江 李亚男 毕晓菲 胡荣锁 陈小爱

关键词:绿咖啡油;微胶囊;复合凝聚法;结构表征;热稳定性
中图分类号:S571.2 文献标识码:A
咖啡是我国重要的热带经济作物,具有极高的经济价值。我国咖啡种植主要分布在云南、海南、四川、台湾等地[1]。据统计,2021 年我国咖啡种植面积已达9.39 万hm2,总产量达10.91 万t[2]。云南省咖啡面积和产量分别占全国的98.84%和99.61%。中国的咖啡消费正在以每年超过15%的速度增长,2021 年中国咖啡市场规模突破1700亿元,居全球第8 位。与咖啡的食用价值相比,作为咖啡衍生产品之一的绿咖啡油(green coffeeoil, GCO)利用率很低,常用于化妆品行业[3]。绿咖啡油富含生育酚和不饱和脂肪酸,如油酸和亚油酸等,可用于生产营养制品[4]。然而其中多不饱和脂肪酸极不稳定且易氧化,导致绿咖啡油保质期缩短及感官、营养品质下降,极大限制了其生物活性的发挥。因此,如何保证绿咖啡油的稳定性,对绿咖啡油的应用具有极其重要的价值。
微胶囊化能使液态的油脂转变为具有亲水性的固体粉末,保护油脂免受外界因素的影响,不仅保持了油脂原有的营养价值,还使其具有稳定的性质和优良的加工特性[5]。微胶囊制备的常用方法包括复合凝聚法、喷雾干燥法和分子包埋法等。韩婕妤等[6]用喷雾干燥法成功制备奇亚籽油微胶囊,有效延缓奇亚籽油的氧化,提高了奇亚籽油的贮藏稳定性。张维等[7]用超声波辅助分子包埋法制备榛子油微胶囊,减缓榛子油氧化速度的同时延长了其货架期。COMUNIAN 等[8]通过喷雾干燥法制备绿咖啡油微胶囊,所制备的微胶囊具有良好的稳定性,具有开发营养食品或化妆品配方的潜力。DE OLIVEIRA 等[9]用复合凝聚法制备绿咖啡油微胶囊并应用到果汁中,绿咖啡油微胶囊可以有效改善果汁流变性,提高感官质量。利用复合凝聚法制备绿咖啡油微胶囊具有操作简单、重现性好、无需复杂设备且制备的产品热稳定性好等优点[9],目前国内外利用复合凝聚法制备绿咖啡油微胶囊的报道较少。
目前复合凝聚技术最常用的壁材是明胶和阿拉伯胶(gum arabic, GA)[10],但牛肉明胶因朊病毒病及其食品安全问题而使其应用范围下降。在食品领域,多种动、植物源蛋白已被开发为明胶的替代品,并与碳水化合物结合后作为复合凝聚微胶囊技术中的壁材[11]。在前期基础中,谭睿等[12]初步评价了不同多糖与明胶组合复合凝聚法包埋绿咖啡油的微胶囊性能,而对动、植物源蛋白与阿拉伯胶为壁材复合凝聚制备绿咖啡油微胶囊还有待研究。
本研究以3种疏水性蛋白和阿拉伯胶为壁材,采用复合凝聚法制备绿咖啡油微胶囊,考察不同壁材对绿咖啡油微胶囊包埋效果、表观形貌、结构组成的影响;探究壁材及其微胶囊内部的相互作用力及热稳定性。该研究为制备绿咖啡油微胶囊提供新的思路和途径,也为绿咖啡油稳定性提升和高值化利用提供理论依据和技术支撑。
1 材料与方法
1.1 材料
1.1.1 材料与试剂 绿咖啡油(C16:0, 5%~20%;C18:0, <7.0%; C18:1, 20%~35%; C18:2, 50%~70%)由雅克耶提芳香医药(青岛)有限公司提供;阿拉伯胶、大豆分离蛋白(soybean proteinisolate, SPI)、尼罗红(用于荧光分析,≥95.0%)、戊二醛为上海阿拉丁生化科技股份有限公司产品;HilmarTM 9410 乳清分离蛋白(whey proteinisolate, WPI; 89%)为美国Hilmar 公司产品;酪蛋白酸钠(sodium caseinate, SC; >90%)、异硫氰酸荧光素(偶联级,90%标记率)、异硫氰酸玫瑰红B(偶联级,70%标记率)为上海源叶生物科技有限公司产品;其他试剂均为国产分析纯。
1.1.2 仪器与设备 Seven Compact S220 pH 计[Mettler Toledo 仪器(上海)有限公司],FJ200-SH型数显高速分散均质机(上海标本模型厂),数显悬臂式恒速强力电动搅拌机(江阴市保利科研器械有限公司),DF-101 集热式恒温加热磁力搅拌器(巩义市予华仪器有限责任公司),SynergyH1全波长扫描式多功能读数仪(美国BioTek 有限公司),Mastersizer 3000 激光粒度仪(英国马尔文仪器有限公司),Phenom Prox 台式显微能谱一体机(荷兰复纳科学仪器有限公司),Nicolet 6700傅里叶变换红外光谱仪(美国Thermo Fisher 公司),FV10i 激光共聚焦显微镜(日本奥林巴斯有限公司),Ultima IV X 射线衍射仪(日本理学株式会社),DSC25 差示扫描量热仪(美国TA公司)。
1.2 方法
1.2.1 浊度的测定 采用大豆分离蛋白(SPI)、乳清分离蛋白(WPI)和酪蛋白酸钠(SC)与阿拉伯胶(GA)分别配制总浓度为0.1%(W/V)的溶液(表1),将溶液置于45 ℃的恒温水浴环境下,逐滴缓慢滴加盐酸(0.1、1.0、10.0 mol/L、),调节混合体系的pH,以0.1 梯度调节pH 至2.0左右,分别测定pH 和波长为600 nm 处吸光度。
1.2.2 复聚物的制备工艺 根据表2 混合均匀制备总生物聚合物浓度为1%的溶液,将混合溶液转移至三口烧瓶,保持体系温度50 ℃,置于恒温加热磁力搅拌器中, 搅拌备用。以攪拌速率400 r/min,在30 min 内缓慢滴加1%或10%盐酸调节乳液的pH 至前述浊度的最优pH。关闭加热源,2 h 内自然冷却至室温后,用冰水浴降温至4 ℃,继续反应1 h。然后将反应体系置于冰水浴中使溶液降温至15 ℃以下保持30 min,以结束复合凝聚反应。用1% NaOH 溶液调节体系pH 至6.0,然后加入5 mL 戊二醛(1%)进行固化,反应时间为3 h。固化后即为绿咖啡油微胶囊悬浮液,静置分层、离心分离上清液后在温度为–80 ℃的超低温冰箱预冻24 h,在–50 ℃和20 kbar 下真空冷冻干燥48 h。所得样品盛放于高密度聚乙烯袋中,并保存于棕色干燥器中备用。
1.2.3 微胶囊的制备工艺 采用复合凝聚法制备绿咖啡油微胶囊(图1)。按表2 分别配置壁材储备液,以芯壁比1∶1 添加绿咖啡油,10 000 r/min均质5 min,得到O/W 乳液;再缓慢加入GA 储备液,10 000 r/min 二次均质5 min,得到生物聚合物浓度为1%的乳液,室温下以500 r/min 速度搅拌,并用1.0 mol/L HCl 缓慢滴加调节pH 至固定值后,凝聚30 min。后续制备过程同1.2.2。
1.2.4 粒度分布 参照YANG 等[13]的方法,采用Mastersizer 3000 激光粒度仪测定绿咖啡油微胶囊的尺寸分布。每个样品重复测定3 次。
1.2.5 微胶囊产率、包埋率的测定 根据以下公式计算微胶囊产率(YE):
微胶囊表面含油量(SO)测定:称取m0 微胶囊粉末,分次加入30 mL 石油醚快速洗涤,然后过滤至恒重圆底烧瓶m1,在45 ℃和100 Mpa压力下进行溶剂回收,收集完毕于恒温干燥箱中干燥1.5 h,冷却称重,重复操作直至恒重m2。根据以下公式计算表面含油量:
微胶囊总含油量(TO)测定:称取1.0 g 微胶囊粉末,用10 mL 蒸馏水(45~50 ℃)分次溶解,加入1.25 mL 浓氨水混匀,置于60 ℃水浴加热5 min,冷却加入10 mL 无水乙醇充分摇匀,再加入25 mL 无水乙醚轻轻振荡1 min 排气,加入25 mL 石油醚剧烈振荡30 s 后静置30 min 分层,待上层澄清时读取上层清液总体积(V0),吸取一定体积的上清液(V1)于已恒重的圆底烧瓶(m1)中。在45 ℃和100 Mpa 压力下进行溶剂回收,收集完毕于恒温干燥箱中干燥1.5 h,冷却称重,重复操作直至恒重m2。根据以下公式计算总含油量:
1.2.6 傅里叶变换红外光谱(FT-IR)分析 参照周麟依等[14]的方法,利用OMNIC 32 软件对光谱进行平滑和基线校正处理,光谱数据输出格式为透光率,重复测定3 次。
1.2.7 扫描电镜(SEM)分析 参照LAN 等[15]的方法,用扫描电镜观察绿咖啡油微胶囊及复聚物的微观形貌。
1.2.8 激光共聚焦扫描显微镜(CLSM)分析 通过激光共聚焦显微镜在双波长下观察复聚物和微胶囊的样品形态。复聚物和微胶囊油滴观察分别采用DUHORANIMANA 等[16]和BAKRY 等[17]的方法。
1.2.9 X 射线衍射分析(XRD) 参照ALI 等[18]的方法,使用Jade 软件下对样品的结晶度进行计算,相对结晶度(relative crystallinity)为衍射峰面积与总衍射峰面积之比。
1.2.10 差示扫描热量法(DSC) 参照SHI 等[19]方法并稍作修改,采用差示扫描量热仪分别对绿咖啡油微胶囊和空囊样品进行扫描,并作差示扫描分析曲线,测定玻璃态转变温度。
1.3 数据处理
采用Origin 96(Northampton, MA, USA)软件绘图,利用SPSS 26.0 软件(IBM Corporation,New York, NY ) 进行样品间的显著性分析(P<0.05)。每个样品重复测定3 次,结果以平均值±标准偏差表示。
2 结果与分析
2.1 pH 及壁材混合比例对不同蛋白质与阿拉伯胶体系浊度的影响
由图2 可知,浊度曲线的起始阶段为体系初始pH(pH 为7.0)下的状态,3 种蛋白质与阿拉伯胶体系浊度均随pH 的减小呈先上升后下降的趋势。当pH 下降至3.0~4.0 时,浊度急速上升至峰值1.20~1.60,溶液呈现清晰的乳白色,并逐渐转向非透明状。浊度跨越峰值后迅速进入下降阶段,与浊度上升过程相比,下降阶段趋势相对平缓且出现拐点,随后进入一个更为迟缓的下降过程。
此外,随着不同蛋白质与阿拉伯胶二者间混合比例的不同,其浊度曲线也存在较大差异。SPI∶GA、WPI∶GA 和SC∶GA 的壁材质量比分别设定为1∶1、2∶1 和2∶1 用于后续微胶囊制备的最优条件,其浊度最高点所对应的pH 3.5、3.7和3.8 分別为相应微胶囊形态的最佳理论值。
2.2 复聚物的结构表征
2.2.1 傅里叶变换红外光谱分析 由图3 可知,1610.27 cm?1和1018.23 cm?1是属于阿拉伯胶的峰值,前者在3 种复聚物的光谱上左移,后者消失。此外,SPI 在3293.82 cm?1处的吸收带于SPIGA 复聚物中移至3208.96 cm?1处,WPI 位于3299.60 cm?1处的吸收带在WPIGA 复聚物中移至3276.40 cm?1,SC 位于3208.96 cm?1处的吸收带在SCGA 复聚物中移至3203.18 cm?1 处。在SPI光谱中,1671.98、1535.05、1247.71 cm?1处的峰对应酰胺Ⅰ、酰胺Ⅱ和酰胺Ⅲ吸收带,分别转移到SPIGA 光谱中1643.05、1536.98、1245.79 cm-1处;在WPI 光谱中,1641.12、1544.70、1249.64 cm-1处的峰对应酰胺Ⅰ、酰胺Ⅱ和酰胺Ⅲ,分别转移到WPIGA 光谱中的1646.91 、1538.91 、1243.86 cm?1 处。
2.2.2 扫描电镜分析 采用扫描电镜对未加入绿咖啡油的干燥凝聚物形态结构进行评价(图4),所有样品图像均显示粉末尺寸较大,形状不规则,呈团聚状存在,也有类似碎玻璃或片状结构。微粒表面较为完整,无明显的裂缝。
2.2.3 激光共聚焦扫描显微镜(CLSM)分析 采用CLSM 观察蛋白质和多糖在复聚物中的分布,蛋白质(SPI/WPI/SC)和阿拉伯胶分别用荧光标记物(FITC 和RBITC)染色。如图5 所示,3 种复聚物大小不规则,凹陷较少且荧光分布均匀。图5A、图5D 和图5G 标记蛋白质显示绿色结构,图5B、图5E 和图5H 标记阿拉伯胶为红色通道,均获得类似的显微图像,表明SPI/WPI/SC 和阿拉伯膠同时存在于凝聚的复聚物中;图5C、图5F和图5I 可以清楚地看到复合凝聚体(黄色)的形成,说明阿拉伯胶与蛋白质紧密结合,其中SCGA颗粒外围有明显的绿色荧光,可能是由于酪蛋白酸钠分子比阿拉伯胶分子密度大造成的。
图片共定位分析常用方法有散点图(图6)、共定位相关系数分析(表3),均可直观说明分子间的相互作用、相对位置关系和空间距离[20]。如图6 所示,WPIGA 的散点图分布呈一条直线,说明2 个荧光通道在成像时强度相当,相关性最强且r=0.945,越趋近于1,共定位程度最高;SPIGA共定位程度次之(r=0.930),而SCGA 的散点图分布偏移对角线,相关性较低(r=0.902),共定位程度最低。共定位分析显示3 种蛋白质与阿拉伯胶分布相似,重合率>90%。
2.3 微胶囊的理化性质
2.3.1 微胶囊的产率及包埋率 由表4 可知,SPIGA-GCO 微胶囊产率最高,平均为86.40%,WPIGA-GCO 的产率最低,SPIGA-GCO、SCGAGCO微胶囊产率与WPIGA-GCO 差异显著(P<0.05)。不同壁材的微胶囊之间包埋率存在显著差异(P<0.05),SPIGA-GCO 的包埋率最高,平均为69.26%,较SCGA-GCO 与WPIGA-GCO具有相对较好的包埋效果。以WPIGA-GCO 的表面含油量最高(15.96%),对应包埋率最低。SCGA-GCO 的TO 含量为三者最高,但包埋率较低。
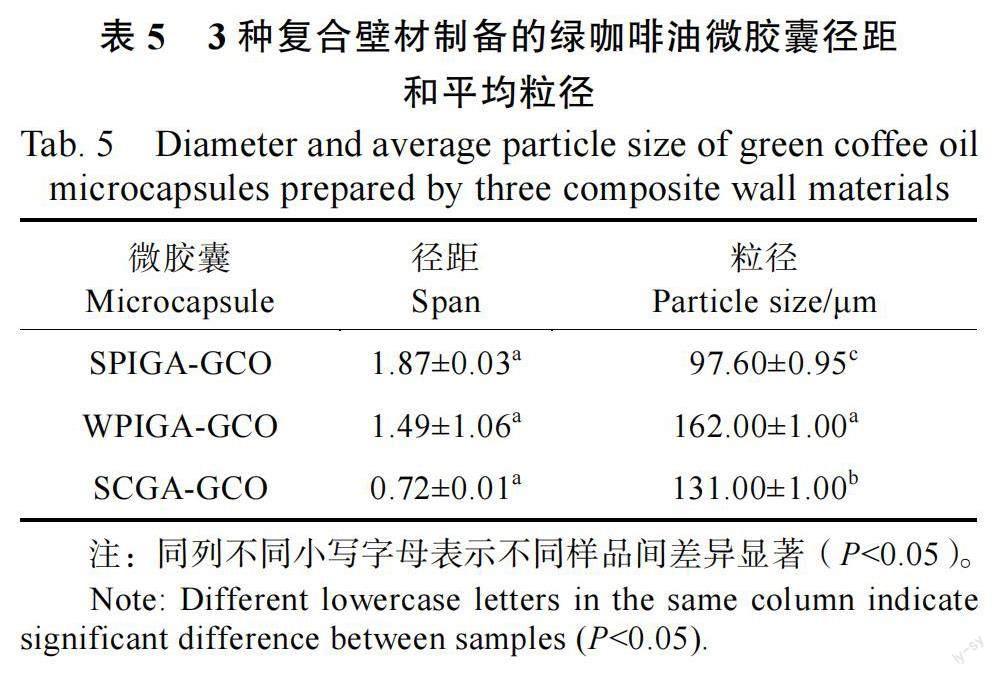
2.3.2 微胶囊的粒径分析 3 种蛋白质与阿拉伯胶为壁材制备的绿咖啡油微胶囊粒径分布均匀,总体上呈正态分布,呈明显的单峰分布(图7)。3 种微胶囊的径距随壁材种类的不同而无明显差异(P≥0.05),范围为0.72~1.87(表5)。壁材种类对绿咖啡油微胶囊平均粒径有显著影响(P<0.05 ), 平均粒径范围97.60~162.00 μm, 其中WPIGA-GCO 的粒径最大(162.00 μm)。
2.4 微胶囊的微观结构分析
2.4.1 FT-IR 分析 绿咖啡油微胶囊的红外光谱如图3 所示。绿咖啡油3464.45 cm–1(O-H 拉伸)和2935.13、2850.27、1745.26 cm–1(C=C 伸缩)呈强振动模式,其中1157.08 cm–1对应C-O-C 伸缩。3 种绿咖啡油微胶囊红外光谱峰位于3542.00~3507.00 cm-1区域内,3 种蛋白质在这个区域也存在类似的峰值范围(3299.00~3208.00 cm–1),是由于O-H 键的伸缩振动,且在GA 粉末中3392.17 cm–1处发现相同峰,为多糖中O-H 相关的典型吸收带伸缩(3550.00~3200.00 cm–1)、脂肪族伯胺(N-H)伸缩(3400.00~3300.00 cm–1)和仲胺(N-H)伸缩(3350.00~3310.00 cm–1)。在游离绿咖啡油的光谱中出现吸收带3465.45 cm–1分别转移到3 种绿咖啡油微胶囊光谱中3131.83、3139.54、3145.32 cm–1处。比较绿咖啡油、3 种复聚物和绿咖啡油微胶囊的光谱, 在2933.00~2850.00 cm-1处出现特征吸收带,证明存在类黄酮的伸缩振动(2920.00~2970.00 cm?1)和亚甲基的不对称振动(2925.00~2853.00 cm?1),表示绿咖啡油已成功封装到3 种不同复聚物中。绿咖啡油光谱中出现1745.26 cm?1吸收峰为甘油三酯羰基官能团强烈特征吸收带,且在3 种微胶囊FT-IR图中1745.00~1743.00 cm?1处也出现其峰值。
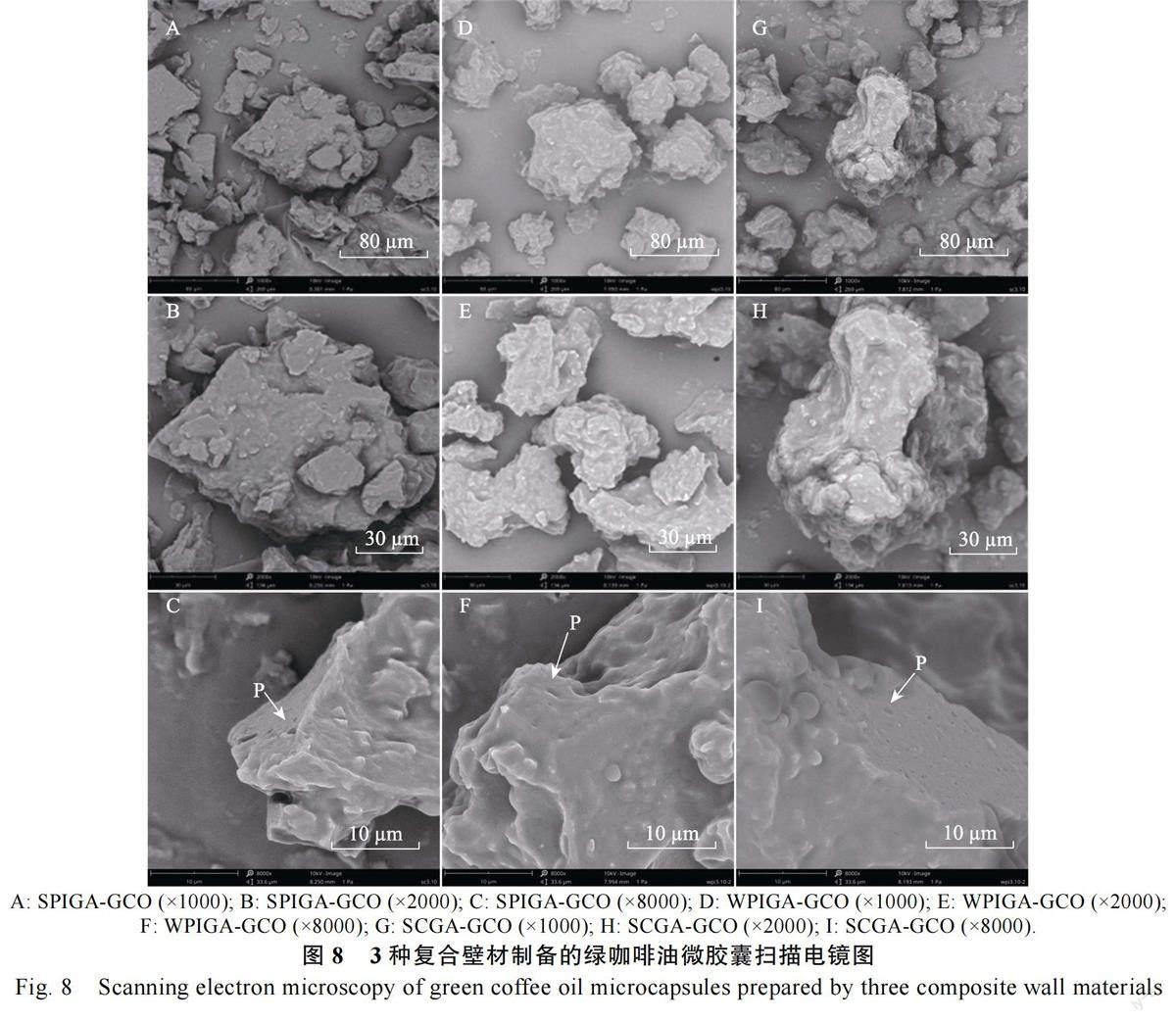
2.4.2 SEM 分析 3 种微胶囊粉末微观结构如图8 所示。所有样品粉末都具有碎玻璃结构和萎缩的纹理,且颗粒形状和大小不同,呈不规则片状结构。SPIGA-GCO、WPIGA-GCO 和SCGA-GCO微胶囊由于蛋白质的乳化特性,油滴在光滑无孔的表面形态下分散,图像显示微粒表面没有裂纹或裂缝的迹象。
2.4.3 CLSM 分析 由图9 可以发现,绿咖啡油微胶囊是不规则粒子,由于CLSM 的景深有限,凹陷表面的扫描盲区显示为黑色。绿色图层(图9A、图9D、图9G)为FITC 所染不同的蛋白质,红色图层(图9B、图9E、图9H)为尼罗红所染的绿咖啡油, 后者的荧光强度反映油量。在WPIGA-GCO和SCGA-GCO颗粒表面有部分强烈的红色荧光,说明表面油量较多且相对包埋率低(图9E、图9H)。SPIGA-GCO 图像显示的荧光强度较低且均匀,表明绿咖啡油被较好地包埋在颗粒内部(图9B)。此外,从图9C、图9F、图9I 中观察到油滴(红色部分)被均匀地嵌入在蛋白质基质(绿色部分)中,无论是外层还是内层,表明绿咖啡油被SPIGA、WPIGA 或SCGA 成功封装。其中SPIGA-GCO 中红色荧光分布更为均匀,说明绿咖啡油被有效包埋在SPIGA 中;而WPIGA-GCO 中部分油滴被组织成小液滴外露在壁材表面(箭头所示)。
2.4.4 XRD 分析 图10 所示,3 种壁材(SPIGA、WPIGA、SCGA)与其绿咖啡油微胶囊的衍射图形大致相似,为一个较宽的漫反射峰,表明绿咖啡油微胶囊具有无定形结构的特征;3 种绿咖啡油微胶囊衍射角(2θ)范围为19.38~20.92°与壁材相比(19.18~19.58°),微胶囊的特征峰都发生了偏移。表明SPIGA-GCO、WPIGA-GCO 和SCGA-GCO 的络合是成功的。此外,3 种壁材与绿咖啡油微胶囊的2θ 附近其峰值銳度和强度存在差距,绿咖啡油微胶囊峰值较窄且强度增加,表明添加绿咖啡油不会显著改变主衍射峰的位置,但会使衍射峰变窄且强度增加。本研究中SPIGA-GCO、WPIGA-GCO 和SCGA-GCO 3 种微胶囊的相对结晶度分别为45.99%、46.37%和42.23%,其中WPIGA-GCO 显示出很小的结晶倾向,与其他壁材相比具有较高的相对结晶度。
2.5 微胶囊的热性能分析
由图11 可知,3 种绿咖啡油微胶囊的玻璃化转变温度均在100 ℃ 以上, SPIGA-GCO 、WPIGA-GCOH 和SCGA-GCO 分别在107.60、108.33、103.93 ℃有明显的吸热峰,而3 种壁材分别在112.12、110.21、109.46 ℃达到热变性温度,三者变化趋势相似,峰位略有移动。
3 讨论
本研究提出了绿咖啡油与3 种蛋白质和阿拉伯胶复聚物作为微胶囊载体的结合条件。
3.1 复聚物最佳pH 和壁材比研究
3种蛋白质与阿拉伯胶体系浊度均随pH 的减小呈先上升后下降的趋势,当pH 下降至3.0~4.0时,浊度急速上升至峰值,浊度跨越峰值后迅速进入较为迟缓的下降阶段。主要是由于随着静电相互作用的增强,先前形成的可溶性复合物发生了结构间的重排,从而形成某种相对紧密的结构。复聚物浊度的峰值还意味着不同蛋白质与阿拉伯胶间的静电相互作用达到了最大值,此刻发生相分离,具备了形成微胶囊的可能性[21]。浊度跨越峰值后迅速进入下降阶段,这可能是由于不可溶性复合物基本消失,体系过渡到可溶性复合物的解体过程中,发生“破乳”现象。不同蛋白质与阿拉伯胶混合比例的不同,其浊度曲线也存在较大差异,说明pH 和壁材质量比都极大地影响了蛋白质和多糖的相互作用以及微胶囊的形成。基于静电相互作用最大即等同于浊度最大的原理,各曲线浊度最大点均为各比例下的静电中和点[22],达到该点可使壁材最大程度被利用。本研究结果与贾聪等[23]和夏慧亭等[24]的报道一致,酪蛋白酸钠和阿拉伯胶的最适pH 与刘春花等[25]的研究结果略有不同,可能是由于在制备蛋白储备液时,盐离子使得后续复合物体系发生改变,最佳条件也会发生细微影响。
3.2 蛋白质与阿拉伯胶制备复聚物的结构表征
3.2.1 傅里叶变换红外光谱(FT-IR)分析 红外光谱是生物聚合物结构分析的有力工具,可提供蛋白质和阿拉伯胶复杂凝聚过程中相互作用的信息[26]。本研究通过FT-IR 扫描图谱和荧光共定位分析证实了蛋白质与阿拉伯胶之间存在静电相互作用,获得了稳定、表观完整的复聚物,为后续的绿咖啡油包埋工作奠定了基础。SPI、WPI 和SC 吸收带在复聚物中发生偏移,表明SPI、WPI和SC 中的O-H 基团与阿拉伯胶中的C=O 基团之间形成了氢键[27]。3 种蛋白质与阿拉伯胶形成复聚物,单一组分的壁材在形成复聚物后在酰胺Ⅰ、Ⅱ和Ⅲ的吸收峰分别有偏移,说明蛋白质的氨基与阿拉伯胶的羧基确实发生静电相互作用。QIU等[28] 通过红外光谱发现绿豆蛋白吸收峰从3294.01 cm-1 移动到绿豆蛋白和杏皮果胶复聚物中的3293.46 cm?1 处,表明绿豆蛋白与杏皮果胶之间发生静电相互作用,与本研究结论一致。
3.2.2 扫描电镜(SEM)分析 SEM 下未加入绿咖啡油的干燥凝聚物微粒表面较为完整,无明显裂缝。说明SPI、WPI、SC 与阿拉伯胶相互作用,构建了致密性良好的壁材体系,促进微胶囊的形成[17]。碳水化合物的添加使得壁材体系在结构形态上更加稳定,对于芯材的保护和避免氧化,提高微胶囊稳定性至关重要。本研究复聚物形态结果与CHARLES 等[29]的研究结果一致,后者发现以麦芽糊精和乳清分离蛋白制备的复聚物其结构不规则,缺乏正常的球形,呈现高度多孔。
3.2.3 激光共聚焦扫描显微镜(CLSM)分析CLSM 下3 种复聚物大小不规则,凹陷较少且荧光分布均匀,这可能是因为碳水化合物组成的壁材体系具有更高的分子柔性,表现出更多的分子构型,导致成膜均匀[18],提高微胶囊稳定性。DUHORANIMANA 等[16]研究表明,蛋白多糖复合凝聚的胶囊壁外部界面上高密度的明胶分子有利于保护芯材。CLSM 分析发现,3 种蛋白质和阿拉伯胶在复聚物内均匀分布,构成稳定的骨架,可赋予微胶囊壁一定的机械强度和致密性,对提高微胶囊的氧化稳定性有重要作用。
3.2.4 共定位分析 不同蛋白和阿拉伯胶复合凝聚后具有良好的相关性,其中WPIGA 相关性最强,说明乳清分离蛋白与阿拉伯胶之间有较强的静电相互作用。本研究发现蛋白质和多糖在颗粒内部均匀分布,构成高稳定性的骨架,共定位分析也显示蛋白与多糖分布相似,重合率达到70%以上,与ZHANG 等[30]的研究结果类似。
3.3 蛋白质与阿拉伯胶制备微胶囊的理化性质、结构及性能特点
3.3.1 理化性质 不同壁材对微胶囊的产率有显著影响,本研究结果与肖军霞等[31]以大豆分离蛋白阿拉伯胶为壁材包埋甜橙油的结果一致。包埋率用来评价粉末状微胶囊内油脂的保护程度,是微胶囊粉末的重要特征。ILYASOGLU 等[32]通过冷冻干燥获得的鱼油微胶囊包埋率为29.40%~81.60%。与本研究所得包埋率相似,说明3 种壁材均有效包埋绿咖啡油。SHAO 等[26]以麦芽糖糊精-蛋白为壁材制备灵芝多糖微胶囊,与大豆分离蛋白和酪蛋白酸钠相比,乳清蛋白和麦芽糖糊精络合制备的微胶囊包埋率达到89.80%,说明适当添加蛋白可改善麦芽糊精的性能,提高包封效率。由此可知,壁材的选择也可改变微胶囊包埋率,且与壁材的种类无线性关系。表面油(SO)表示存在于微胶囊表面未被包埋的绿咖啡油,易发生氧化变质,从而降低功能成分的保质期[33],是导致包埋率较低的主要因素之一。因此应尽量减少脂质封装中的表面油量。WPIGA-GCO 的SO 较高,对应包埋率较低。SCGA-GCO 的SO 含量为最高,但包埋率较低,由此可知,尽管粉末中的高含油量是理想的,但相对较高的包埋率也可能导致芯材损失[34],因此还需最大限度保护油的同时减少表面油含量。
3.3.2 粒径 粒径大小是微胶囊应用于食品基质的一个主要特征。壁材种类对绿咖啡油微胶囊平均粒径有显著影响,本研究表明WPIGA-GCO 的粒径最大,可能是由于WPI 的乳化和成膜能力在干燥过程中诱导微胶囊膨胀或膨化,通过增加封闭空气含量来增加粒径尺寸。这一结果与石泽栋等[10]对牛至精油微胶囊化的结果相似,其微胶囊平均粒径为147.00 μm。
3.3.3 FT-IR FT-IR 是生物大分子及其聚合物结构分析的有力工具,通过FI-TR 扫描分析,可以显示各物质的分子结构和特征化学键[35],可以证明微胶囊的形成以及物质之间是否存在相互作用。绿咖啡油微胶囊的红外光谱显示,绿咖啡油具有强烈的疏水性[36],壁材和芯材之间发生氢键效应,对应酯的存在[28],绿咖啡油成功包埋于3种复聚物中,且与壁材未发生化学反应。
3.3.4 微观结构 SEM 显示3 种微胶囊粉末微观结构,显示微粒表面无裂纹或裂缝的迹象,但是不同微胶囊截面存在相似的微腔和孔隙,可能是由于乳状液在冷冻过程中形成较小的冰晶,且在冷冻干燥过程中升华,从而形成多孔结构。YANG等[37]研究解释了微胶囊表面光滑是因为蛋白质构象会随着蛋白质与不同材料的相互作用而改变,趋向于更有利的相互作用,如通过静电相互作用能够稳定复合物和降低系统的表面张力。本研究结果显示,3 种配方制备的绿咖啡油微胶囊壁结构相对完整,由此可推断微胶囊对芯材具有良好的保护作用。此外,实验结果与其表面油含量和包埋率的测定结果一致,以SPI 和阿拉伯胶为壁材制备的绿咖啡油微胶囊表面最平整光滑,其表面油含量最低,包埋率最好。SEM 显示了绿咖啡油微胶囊的表观形态,但不能直观地观察到微胶囊的油滴分布,因此,使用多重标记的CLSM观察3 种绿咖啡油微胶囊中油滴分布,CLSM 图像证实了绿咖啡油已成功包埋在3 种复合壁材中,且印证了SEM 图像的观察结果。这一结果进一步说明SPIGA、WPIGA 和SCGA 可作为包埋绿咖啡油的微胶囊壁材。
3.3.5 XRD为了研究复合壁材和绿咖啡油微胶囊的晶体结构及其结晶度进行了XRD 测定。一般来说,晶体材料的峰较尖,而非晶材料的峰较宽。本研究显示,芯材和壁材之间具有高度相容性,且包埋后不会在很大程度上破坏壁材的无定形结构,说明绿咖啡油微胶囊已经形成,与曹俊英等[38]的研究结果一致。一般来说,无定形壁材比结晶壁材具有更强的吸湿性和水溶性,因此在贮藏过程中会吸附更多的水,并通过微观结构的破坏、营养物质的降解和微生物的不稳定性降低了微胶囊的贮藏稳定性。为了研究绿咖啡油微胶囊的稳定性,计算了微胶囊的结晶度。本研究中SPIGA-GCO、WPIGA-GCO 和SCGA-GCO 3 种微胶囊的相对结晶度分别为45.99%、46.37%和42.23%,表明该晶体结构是理想的,所得结果与ALI 等[18]报道苦参提取物微胶囊的相对结晶度(34.03%~46.52%)类似。其中WPIGA-GCO 显示出很小的结晶倾向,与其他壁材相比具有较高的相对结晶度,可能是由于分子间相互作用,结晶度和游离体积均会影响势垒的渗透率,具有相对较好的贮藏稳定性。
3.3.6 热性能 采用DSC 测定样品在控温过程中吸收或释放的热量,以评价样品的热力学性质。在DSC 测量中,热分析谱上的吸热峰代表了试样的热变性温度区,该峰值对应的温度即为热变性温度(玻璃化转变温度),样品的变性温度越高,热稳定性越高[39]。本研究中,3 种绿咖啡油微胶囊的玻璃化转变温度均在100 ℃以上,说明微胶囊化对绿咖啡油具有良好的保护作用。SPIGA-GCO、WPIGA-GCOH 和SCGA-GCO 出现明显的吸热峰与达到热变性温度时二者趋势相似,峰位略有移动,其原因可能是2 种体系的孔隙率和微观结构的差异。BURHAN 等[39]制备的肉桂精油纳米微胶囊具有较高的热稳定性,发现高热稳定性与蛋白质和多糖分子间的相互作用有关,与本研究结果相似。
综上,微胶囊实验表明,不同蛋白质与阿拉伯胶的组合对微胶囊的粒径和包埋率有显著影响;通过傅里叶红外光谱发现壁材和芯材之间存在较强的氢键效应;SEM 和CLSM 表征结果证明绿咖啡油最终包埋在3 种壁材中;XRD 图谱表明3 种微胶囊具有无定形结构,且绿咖啡油的嵌入对微胶囊的结晶状态无显著影响;通过DSC 对不同蛋白质与阿拉伯胶形成的复聚物热稳定性进行分析,得出其微胶囊产品性质比较稳定,整体结构完整,表明3 种复合壁材可对绿咖啡油有良好的保护作用。其中SPIGA-GCO 的产率最高(86.40%)、粒径最小(97.60 μm)、包封率最大(69.26%)、油滴能夠均匀嵌入壁材并具有更好的热稳定性(107.60 ℃),表明用大豆分离蛋白和阿拉伯胶对绿咖啡油包埋效果更优,更有利于绿咖啡油的储存和使用。以3 种疏水蛋白质与阿拉伯胶为壁材,通过复合凝聚法制备绿咖啡油微胶囊具有可行性,所制备的绿咖啡油微胶囊表现出优良的热稳定性,为绿咖啡油在食品领域的深加工利用提供了理论依据和技术支撑。
