Bottom-up design and assembly with superatomic building blocks
2022-12-28FaminYu于法民ZhonghuaLiu刘中华JiaruiLi李佳芮WanrongHuang黄婉蓉XinruiYang杨欣瑞andZhigangWang王志刚
Famin Yu(于法民) Zhonghua Liu(刘中华) Jiarui Li(李佳芮) Wanrong Huang(黄婉蓉)Xinrui Yang(杨欣瑞) and Zhigang Wang(王志刚)
1Institute of Atomic and Molecular Physics,Jilin University,Changchun 130012,China
2College of Physics,Jilin University,Changchun 130012,China
3Institute of Theoretical Chemistry,College of Chemistry,Jilin University,Changchun 130023,China
Keywords: superatom,bottom-up,assembly,atomic level
1. Introduction
Designing specific atomically precise materials has attracted much attention in recent years,[1–4]including the organic,[5,6]metallic,[7,8]alloyed,[9]rare-earth,[10]and metalorganic frameworks,[11]etc.[12]However, the traditional topdown approaches (e.g., physical mechanical exfoliation and etching) suffer from the limitations that it is difficult to control microscopic morphology and may destroy the original properties of units.[13–15]In 1960,Feynman proposed that the quantum confinement effect of electrons enabled nanoparticles to exhibit uncommon properties.[16]It can be concluded that there is plenty of room at the bottom to synthesize novel materials,and the bottom-up design approach is currently attracting a lot of attention.[17,18]Not only that,since the structures that can be used as the bottom units of material science far exceed the types of elements in the periodic table,the development of the bottom-up atomic-level approach that directly targets specific needs will also promote a new paradigm,[19–21]which is of great significance for the future.
Superatoms originate from cluster structures composed of atoms, and they have discrete superatomic molecular orbitals(SAMOs) that exhibit symmetries like atomic orbitals.[22–27]Consequently,superatoms are ideal atomic-level bottom units that meet Feynman’s expectations, and bottom-up assembly based on superatoms has become an important research direction.[9,28–30]Among the superatoms,it was found that the light actinide(An)elements Ac, Th, Pa, U and Pu embedded in fullerene C28can form a series of stable endohedral metallofullerene(EMF)superatomic structures with gradual electron arrangement.[31–35]In particular,the two unpaired electrons on the C28cage facilitate bonding with neighboring units by spinpolarized magnetic coupling.[36]Thus, actinide-based EMF superatoms have the potential to serve as artificial units for the bottom-up design of desirable structures with known bonding properties. Recently, a planar single-crystal fullerene was reported in which C60cages are covalently bonded with each other.[37]Evidently,artificial fullerenes are potential units for building unique planar topology structures. Despite the above understanding, assembly as a specific requirement generally faces complex intra-and inter-molecular interactions and still requires a clearer understanding of the path from the unit design to assembly.
In this work,we took a series of actinide-based EMF superatoms An@C28(An=Ac,Pa,U,Np and Pu)as the units,on the Au(111)surface,to realize a complex assembly system including a rich variety of one, two, three and four chemical bonding, as well as including the intermolecular dispersion adsorption between different parts. The distinction in this study is that,unlike previous large-scale structural searches for possible building blocks, we first conceived a complex system containing rich intra- and inter-molecular interactions as a specific requirement. Then,based on the different electronic structures of EMF superatoms obtained at the atomic level, a complex structure with different bonding properties was constructed on the Au(111)surface.Therefore,this study not only develops a bottom-up assembly strategy based on the superatomic artificial units but also contributes to the establishment of a new research paradigm.
2. Methods
To carry out this research,the third-generation dispersioncorrected density functional theory (DFT-D3)[38]of firstprinciples was used in optimizing the structures of An@C28(An=Ac,Pa,U,Np and Pu). Specifically,the functional used was Perdew–Burke–Ernzerhof(PBE),[39]which has proved to have an advantage in the calculation of actinide-based EMF superatoms.[33,35]The double-ζbasis set was used for C.[40]In terms of the relativistic effect, Stuttgart–Dresden (SDD)pseudopotential and basis set were used for actinide atoms(ECP60MWB for Ac,Pa,U,Np and Pu).[41]
The Au(111) surface was selected as the substrate because the fullerene can be physically adsorbed on the Au surface.[42]Moreover, the charge transfer between them is negligible, and it is more likely to detach than adsorbed on Ag and Cu surfaces.[43,44]Further, to study the bulk complex located on the Au substrate, quantum mechanics/molecular mechanics (QM/MM) simulations were performed,[45]using the ONIOM (our Own N-layer Integrated molecular Orbital molecular Mechanics)method.[46]It has been applied in many important chemical, biological, and material systems.[47]In this work, the QM region for the complex was treated at the PBE-D3 level with SDD pseudopotential and basis set(ECP60MWB for Ac, Pa, U, Np and Pu) for actinide atoms while the double-ζbasis set for C.The MM region was frozen and was described using the universal force field(UFF),which was developed to provide a reliable description for bonded and nonbonded interactions systems containing all the periodic table elements.[48]No symmetry restriction was imposed on the optimization calculation. The Gaussian 16 package[49]was used to optimize the geometric structures while the Multiwfn 3.8 package[50]was used to analyse the electronic structures.
3. Results and discussion
By embedding actinide atoms (including Ac, Pa, U, Np and Pu) into the C28cage, a series of EMF superatoms Ac@C28, Pa@C28, U@C28, Np@C28and Pu@C28superatoms were formed (Fig. 1(a)). Different from the atomembedded C60structure that loses the symmetry due to the off-center position of the embedded atom, the embedded atoms of An@C28are located at the center and maintain high symmetry.[51,52]The high symmetry,similar size and structure of An@C28help to assemble each other. The electronic structure of these EMF superatoms also follows the same rules.The valence electrons of actinide atoms(3 for Ac,5 for Pa,6 for U,7 for Np,and 8 for Pu)add the 28 valence electrons of C28up to 31,33,34,35,and 36,respectively. Their valence electrons first adopt the 32-electronic rule, and the remaining unpaired electrons are used to form chemical bonds (Fig. 1(b)). The An@C28following the 32-electronic rule is more stable than hollow C28.[35]Among them,Ac@C28and Pa@C28with one unpaired electron can form one covalent bond by sharing an electron. U@C28,Np@C28and Pu@C28have two,three and four unpaired electrons, respectively, resulting in the formation of the corresponding number of covalent bonds by sharing electrons(see Figs.S1–S5 of supporting information (SI)for details). Hence, actinide-based EMF superatoms are promising candidates for building controllable structures.
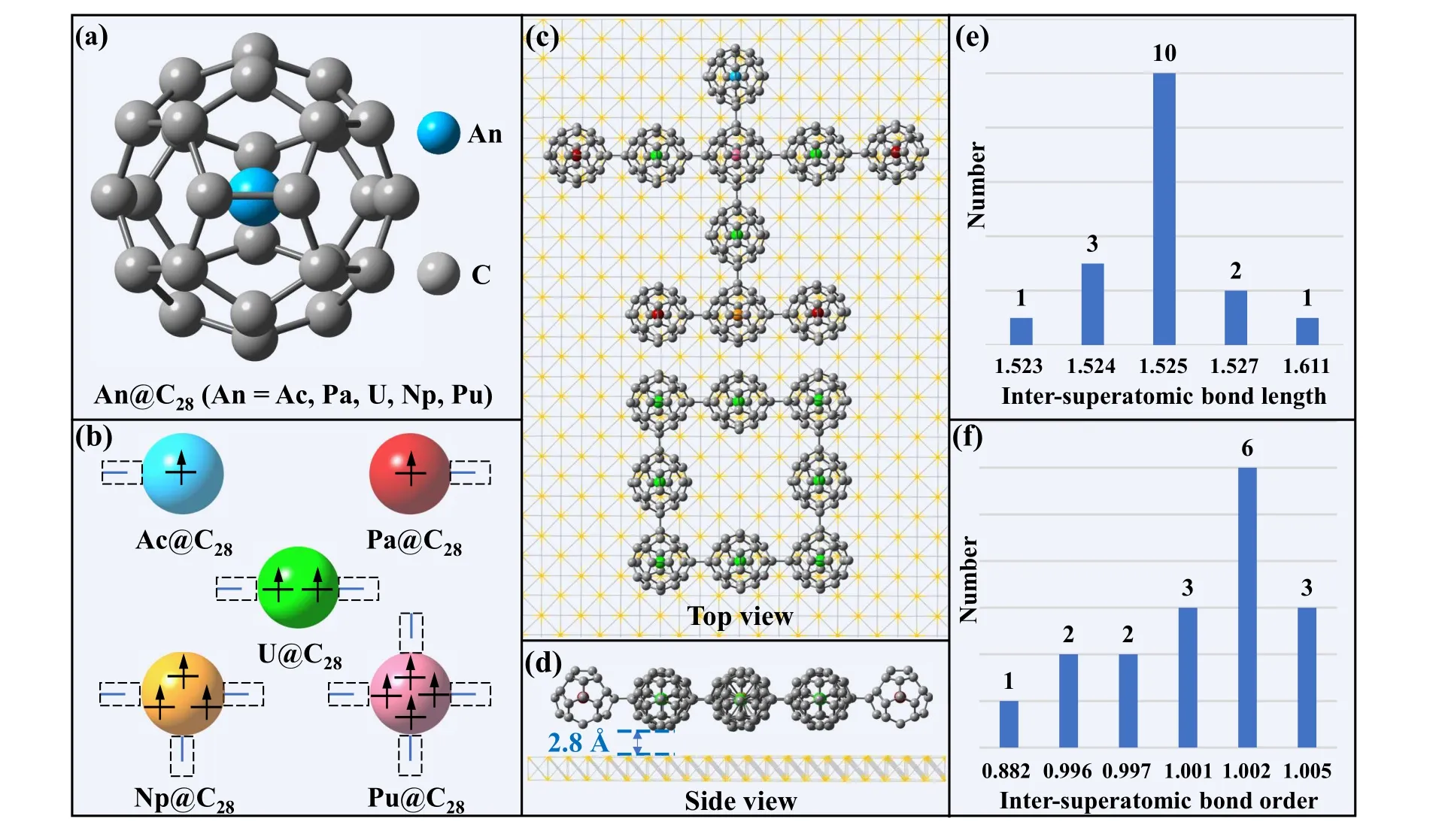
Fig.1. Structural analyses of the complex assembled by EMF superatoms. (a)and(b)The structural representation and the number of chemical bonds that can be formed for An@C28 (An=Ac,Pa,U,Np and Pu),respectively. (c)and(d)The top and side views of the assembled complex. Color codes:grew,C;blue,Ac;red,Pa;green,U;orange,Np;pink,Pu. The blue label is the distance between the complex and the substrate. (e)and(f)The bond length and bond order analyses of inter-superatomic C–C bonds,respectively.
Based on the above discussions, we designed a bulk planar complex on the Au(111) surface. For the optimized structure, the distance between An@C28and Au substrate is 2.8±0.1 ˚A (Figs. 1(c) and 1(d)), which is consist with the result of a low-energy electron diffraction experiment.[42]Obviously,this has exceeded the bond length of the Au–C single bond(2.0–2.3 ˚A),[53]and belongs to physical adsorption. The distance of the complex from the substrate boundary is sufficiently far to avoid boundary effects (see Fig. S6 of SI). In the complex, there are many different interactions, including one,two,three,and four chemical bonds for a superatom and weak interactions between closed-shell parts. Hence, multiple analyses were performed to confirm that the interactions in the complex are as expected. First, to verify that the number of inter-molecular chemical bonds is determined by the number of valence electrons,the bond length and Mayer bond order analyses were performed. The results show that the inter-superatomic C–C bond length is between 1.523–1.611 ˚A and the bond order of these chemical bonds is close to 1(Figs. 1(e) and 1(f)), indicating that these bonds are covalent bonds formed by sharing electrons between superatoms.Hence, the number of 17 inter-molecular covalent bonds corresponds exactly to the 34 unpaired electrons in the whole system. Orbital analysis of the complex suggests that the units are connected by valence orbital fusion (see Fig. S7 of SI).In addition,the upper and lower parts are connected by weak interactions,since the shortest bond length between the superatoms in the upper and lower parts is about 3 ˚A and the corresponding bond order is zero. Therefore, predictable intersuperatomic interaction systems can be constructed from the bottom up based on the understanding of the bonding properties of the superatomic units.
The electron density difference(EDD)and charge population analyses also prove that the number of inter-molecular chemical bonds is determined by the number of valence electrons. These analyses reveal only a small amount of charge transfer between superatoms in the upper part and no charge transfer between superatoms in the lower part, based on each unit being divided into a fragment (see Fig. S8 of SI). This indicates that they are bonded by sharing unpaired electrons.In addition,the upper part of the complex is composed of five kinds of superatoms and staggered connections. The charge transfer between different superatoms suggests that the chemical bonds between different superatoms are polar. Moreover,the lower part of the complex assembled by the same units has no charge transfer between them, indicating the formation of non-polar inter-molecular bonds.This conclusion is consistent with previous studies showing that polar bonds are formed between heterodimers,and non-polar bonds are formed between homodimers.[54,55]
To investigate the weak interaction in the complex, the upper and lower parts were divided into two fragments(Fig. 2(a)). It is shown that there is no charge transfer between the two fragments. Combining the analyses of bond length and Mayer bond order, they both prove that the upper and lower parts are connected by weak interactions. Furthermore, the van der Waals (vdW) surface is around the complex rather than cutting off the upper and lower parts,showing electron density between them is more than 0.001. This also proves that there is a weak interaction between them.
The independent gradient model based on Hirshfeld partition of molecular density (IGMH) analysis was performed to further visualize the weak interaction in the complex(Fig. 2(b)).[56]In the calculation, the upper and lower parts were defined as individual fragments. In the IGMH map, the map function sign(λ)ρ ≈0 represents the vdW interaction,the area of sign(λ)ρ<0 represents the attractive weak interaction, and the area of sign(λ)ρ>0 represents the repulsive interaction. It can be seen that a thin and broad isosurface appears between the fragments. The isosurface defined by the IGMH exhibits weak interaction. The light green region between the upper and lower parts is commonly dispersion interaction.This results in the weak interaction between the closedshell upper and lower parts.

Fig.2. The interaction analysis of the complex. (a)The electron density difference analysis between the upper and lower parts. The blue line is the van der Waals(vdW)surface,the electron density is 0.001. (b)The sign(λ2)colored scatter map,and the isosurface corresponds to the independent gradient model based on Hirshfeld partition of molecular density (IGMH) analysis of the complex. The correspondence between the peaks and maximum of δginter in the isosurface map is indicated via green arrows.
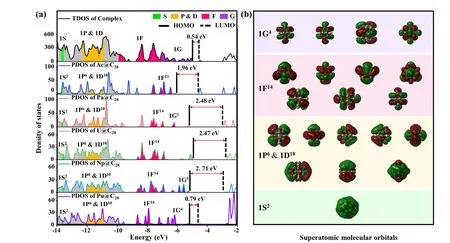
Fig. 3. Electronic structures of the complex and its building blocks An@C28 (An=Ac, Pa, U, Np and Pu). (a) The TDOS of the complex and the PDOS of An@C28 in the complex. The vertical black line and the dotted line indicate the locations of HOMO and LUMO,and the values represent the HOMO–LUMO gap. The green,yellow,pink,and purple regions correspond to S,P&D,F and G SAMOs,and the grey and white regions represent electron occupancy and no electron occupancy. (b)Electrons occupied SAMOs of typical structure Pu@C28. Here,the isosurface is 0.01.
To explore the relation of chemical properties between the complex and units, the total density of states (TDOS) of the complex and particle density of states(PDOS)of units in the complex were calculated (Fig. 3(a)). Due to the Pu@C28superatom has more valence electrons than other building blocks,its electrons occupied SAMOs are shown as typical orbitals(Fig.3(b)). Compared to the PDOS of units,the TDOS curve of the complex presents a smooth trend and smaller gap between the highest occupied molecular orbital(HOMO)and the lowest unoccupied molecular orbital (LUMO), caused by the increase in the number of electrons. The PDOS curves for the EMF units present similar trends,with the differences mainly around the frontier molecular orbitals,due to the different number of valence electrons. In addition,the curve frames of both SAMOs and non-SAMOs are reflected in the TDOS curves of the complexes. Similar trends in the DOS curves of the complex and units suggest that they have similar chemical properties. Therefore,EMF superatoms with different central atoms from the same period can not only control the number of bonds but also build complex structures with similar chemical properties. Evidently,the bottom-up approach shows great potential in the field of accurate assembly while preserving the properties of the units.
4. Conclusion
In summary, we have demonstrated that the pathway for constructing the demanding structures can be achieved by artificial superatomic units assembled from the bottom up. The mechanism of this precise assembly strategy is that the bonding properties can be regulated by adjusting central atoms in An@C28(An = Ac, Pa, U, Np and Pu), such as the formation of one,two,three and four chemical bonds or weak interactions between superatoms. Obviously, since our structures are microscopically at atomic-level precision, the properties of the constructed system are explicitly governed by quantum mechanics. Different from previous large-scale structural searches for possible assembled systems, we first propose a new paradigm for constructing the complex system with rich intra- and inter-molecular interactions based on the obtained superatoms with different kinds of electronic structures. In contrast to the top-down approach, the bottom-up approach has the advantage of high precision at the atomic level and forming materials or devices different from those composed of atoms. Therefore, this work provides a reference for designing demand materials from the bottom up and will facilitate the development of the new paradigm at the atomic level.
Acknowledgments
This work is dedicated to the 70th anniversary of physics and chemistry at Jilin University.
This work is supported by the National Natural Science Foundation of China (Grant Nos. 11974136, 11674123, and 11374004). Z. W. also acknowledges the High-Performance Computing Center of Jilin University and National Supercomputing Center in Shanghai.
猜你喜欢
杂志排行

Chinese Physics B的其它文章
- Editorial:Celebrating the 30 Wonderful Year Journey of Chinese Physics B
- Attosecond spectroscopy for filming the ultrafast movies of atoms,molecules and solids
- Advances of phononics in 20122022
- A sport and a pastime: Model design and computation in quantum many-body systems
- Molecular beam epitaxy growth of quantum devices
- Single-molecular methodologies for the physical biology of protein machines