替比培南及其侧链的合成研究概况
2022-09-14马巧巧王琛彭久合吴晓阳
*马巧巧 王琛 彭久合 吴晓阳
(天津市敬业精细化工有限公司 天津 300450)
替比培南匹伏酯又称泰比培南酯(Tebipenem Pivoxil),化学名称(4R,5S,6S)-6-(特戊酰氧)甲基-3-((1-(4,5-二氢噻吩啉-2基)吖丁啶-3-基)巯基)-6((R)-1-羟乙基)-4-甲基-7-氧-1-氮杂双环[3.2.0]庚烷-2-烯-2-羧酸酯。由日本惠氏立达公司研究开发[1],于2002年转让给日本明治制药株式会社,2009年上市,是目前第一个可以通过口服入药的碳青霉烯类抗生素。由于毒性较低,可作为儿童用药[2-5]。临床适应症为儿童感染的肺炎、中耳炎和鼻窦炎等[6,7]。替比培南匹伏酯抗菌谱广,对大多数临床分离的菌株均表现出比青霉素系列及头孢系列更强的抗菌活性,而与其它注射用的培南类抗生素相比,替比培南匹伏酯也表现出相同或更好的抗菌效果[8-11]。
因替比培南类药物2009年才于日本上市,目前国内尚无厂家生产,也未看到确切研发成功的报道。替比培南匹伏酯的合成可分为三部分,替比培南侧链,替比培南母核(MAP)以及替比培南酯的合成。母核MAP的合成研究较多,工艺成熟,因此,替比培南酯的合成关键在于替比培南侧链的工艺研究[5]。
替比培南中间体侧链,系统命名为1-(4,5-二氢-2-噻唑啉基)-3-巯基吖丁啶盐酸盐,其结构中含有的四元氮杂环,稳定性差,易分解,对酸碱度、温度等反应条件较敏感,目前对其合成路线报道的文献较少,国内鲜有厂家进行研究生产。
1.替比培南侧链研究现状与合成路线
替比培南于2009年在日本上市,最初启动该项目也是日本人。而作为替比培南匹伏酯合成的关键,针对于替比培南侧链的专门研究,国内外报道较少。
目前,从可以查阅到的文献专利来看,替比培南匹伏酯侧链合成的先进工艺及关键技术的知识产权均为日本人所有,主要是Kazuhiko团队和Takeshi Isoda团队。Kazuhiko团队开发了替比培南侧链两条合成路线[1,7],并申请了相关专利US5659043[25]、US5783703[26]、EP717042[27]。Takeshi Isoda 团队对替比培南侧链的三条合成路线进行了报道[8-10],发表的相关专利有EP632039、JP09110868、JP09110869、WO9721712、JP2002003491、JP2002003490、JP2000355592。常州锐博生物科技有限公司公开了一种以苯甲醛和环氧氯丙烷为起始原料,避开高压脱苄基的缺陷的合成方法,发表的相关专利为CN 201310302734.0[12]。
综合上述文献,发现替比培南侧链的合成可概括为6条合成路线,前5条路线由日本人优化得到[2-5],第6条是我国研究人员魏峰等人提出[12]。
合成路线一:
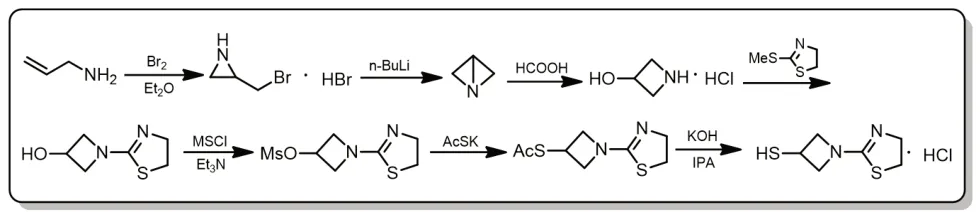
图1 替比培南侧链合成路线一
Kazuhiko等[1,6,25-27]以烯丙胺和溴为起始原料经七步反应制备替比培南侧链,本路线的关键中间体为1-氮杂双环[1.1.0]丁烷,该物质内张力大,不稳定易变质,且制备条件苛刻,需在-78℃,用BuLi合成,难以实现工业化生产。
合成路线二:
路线二仍以烯丙胺和溴单质为起始原料,是Kazuhiko[1,6,25-27]团队在路线一的基础上进行的优化。该路线中采用关键中间体1-氮杂双环[1.1.0]丁烷与巯基乙酸作用,水解直接得到3-巯基吖啶盐酸盐,再与中间体2-甲硫基-2-噻唑啉反应得到替比培南侧链。且Kazuhiko等人[1,6,25-27]还在相关专利中提供了通过烯丙胺与二氯亚砜制备2-氯甲基吖啶盐酸盐,可替代2-溴甲基吖啶溴化氢盐的方法。本路线尽管简化了操作过程,提升了收率,但仍需采用苛刻条件制备中间体1-氮杂双环[1.1.0]丁烷。(注:Kazuhiko等人还制备了替比培南的甲磺酸盐。)
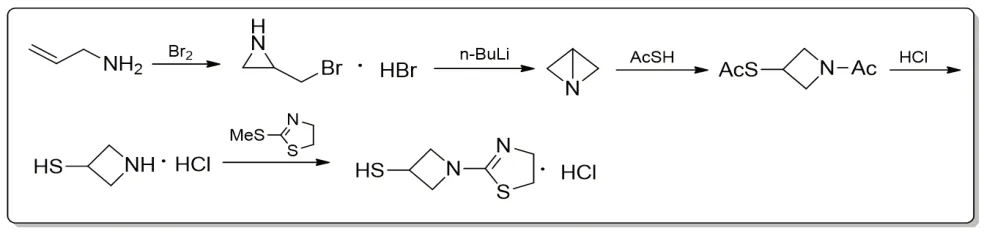
图2 替比培南侧链合成路线二
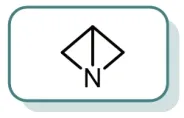
图3 1-氮杂双环[1.1.0]丁烷
1-氮杂双环[1.1.0]丁烷,是替比培南侧链合成的关键中间体,但其内张力较大,极不稳定。目前仅发现3篇文献对它的合成进行了专门报道。1969年,Funke等[28-29]首次报道采用二溴代物的胺盐进行制备,但所用试剂昂贵,且仅以7%的收率得到;1995年Paritosh[1,6]以环氧氯丙烷为起始原料可通过复杂的制备过程以不错的收率得到;1998年Bartnik and Cal通过便宜易得的烯丙胺一锅法制备了1-氮杂双环[1.1.0]丁烷;1999年Kazuhiko[1]对以烯丙胺制备的方法进行了优化,可以60%以上收率一锅法直接制备得到3-羟基吖啶盐酸盐等替比培南侧链相关的中间体。
合成路线三:
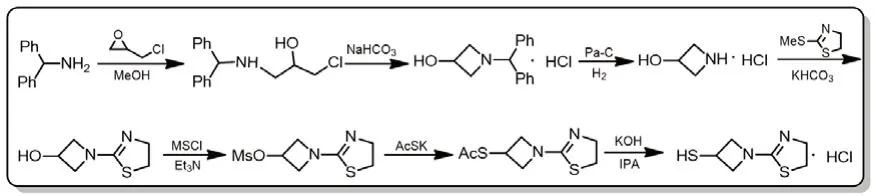
图4 替比培南侧链合成路线三
Takeshi Isoda等在2006年以二苯甲胺与环氧氯丙烷为起始原料,先通过缩合、环化、Pa-C氢气还原得到关键中间体3-羟基吖啶盐酸盐,与中间体2-甲硫基-2-噻唑啉反应后,经甲磺酰化、硫代醋酸钾取代,水解共七步反应,总收率31%,得目标化合物替比培南侧链。同年,Takeshi Isoda等人[8-11]报道中间体3-乙酰硫基-1-(1,3-噻唑林-2-基)吖丁啶成油状物不适合大规模生产,因此又设计了路线四、五。
合成路线四:
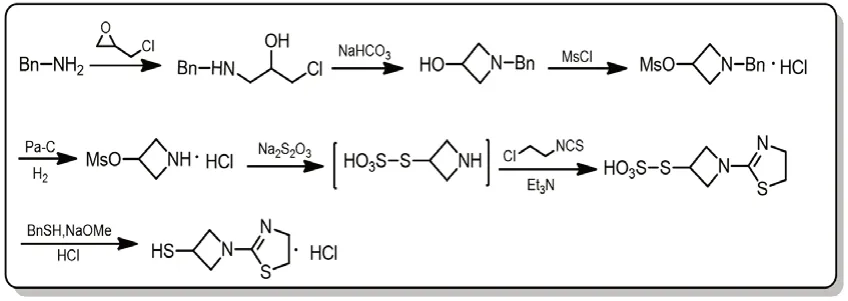
图5 替比培南侧链合成路线四
该路线以苄胺和环氧氯丙烷为起始原料,经缩合、环化、甲磺酰化、Pa-C氢气还原得到关键中间体3-甲磺酰氧基吖啶盐酸盐后,以72%的收率一锅法制备1-(1,3-噻唑啉-2-基)氮杂环丁基-3-硫代磺酸。首先采用Bunte Salt法以磺酰基代替乙酰基保护巯基,再与异硫氰酸氯代乙酯形成噻唑啉环。Bunte Salt法中用到的硫代硫酸钠便宜易得,Takeshi Isoda等[8-11]通过该方法已实现公斤级规模的生产。但在本路线中最后一步去除保护基的过程中,易产生10%左右的二硫代杂质,难以分离,因此设计了路线五避免该杂质的残留。
合成路线五:
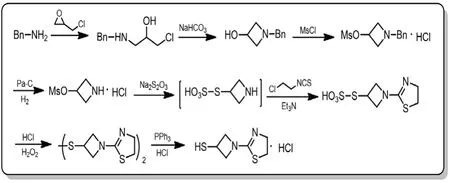
图6 替比培南侧链合成路线五
路线五与路线四的最大不同在于,在制备得到中间体1-(1,3-噻唑啉-2-基)氮杂环丁基-3-硫代磺酸后,采用双氧水氧化将其完全转化为二硫代物,再经三苯基膦还原以高纯度、高收率得到1-(4,5-二氢-2-噻唑啉基)-3-巯基吖丁啶盐酸盐[8-11]。
合成路线六:
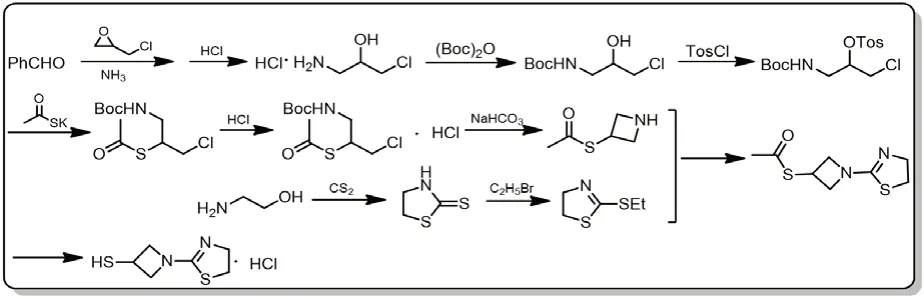
图7 替比培南侧链合成路线六
路线六则以苯甲醛与环氧氯丙烷为起始原料,采用Boc保护氨基,在合成N-杂环丁烷衍生物时避开了传统方法中使用高压脱除苄基的缺陷,安全操作性高。但该路线较长,目前是否应用于实际生产尚未可知[12]。
2.替比培南匹伏酯研究现状
在替比培南匹伏酯相关文献中,国内大部分专利均针对于替比培南匹伏酯的制备(母核与侧链的对接)及其纯化方法,几乎没有专门研究替比培南侧链的合成方法的文献。国内医药公司更多致力于替比培南匹伏酯制备方法的优化与创新,并拥有相关知识产权。
上海医药工业研究院于2011年公开了一种培南母核与替比培南侧链的二聚物直接反应制备替比培南匹伏酯的方法,防止替比培南侧链制备过程中产生无法除去的氧化产物残留于终产品中[13];同年,对采用一种或多种的钯类、镍类和铂类催化剂催化氢化替比培南匹伏酯前体中羧基保护基的制备方法,申请了专利保护[14]。
深圳万乐药业有限公司开发了一种适合工业化生产替比培南的方法,该方法相比于现有的合成替比培南的方法使用的有机溶剂单一,便于回收利用,合成得到的替比培南粗品纯度达到99.8%以上[15]。该公司采用丙酮和异丙醚进行替比培南匹伏酯重结晶精制,操作简单,无需加热或降温设备,室温即可析晶,重结晶收率高[16]。
2012年,江苏济川制药有限公司提供了一种采用替比培南四水合物、碘盐和二异丙基乙胺和特戊酸氯甲酯在溶剂中反应,改进替比培南匹伏酯的工艺[17]。凌沛学[18]则发明了替比培南酯的中间体A型结晶制备方法以及由A型结晶转晶制备B型结晶的方法。
高瑞耀业(北京)科技有限公司在乙醇、乙腈、乙酸乙酯、丙酮、正己烷的单一或混合溶剂中进行溶解、重结晶,制备出了高纯度的替比培南匹伏酯的结晶物[19]。
2013年,山东罗欣药业股份有限公司在制备替比培南过程中依据反应机理优化了溶剂,改进了纯化操作,提高了替比培南匹伏酯的收率[20]。而山东鲁南制药集团股份有限公司也于同年,将替比培南、溶剂Ⅰ、盐、相转移催化剂混合,常温进行成盐反应,然后加入特戊酸氯甲酯进行酯化反应,反应完毕,提取,浓缩,滴加溶剂Ⅱ析晶,过滤得替比培南酯,纯度达到99.7%以上,无需进一步精制[21]。
2016年,河南全宇制药股份有限公司不仅提供了一种明显提高中间体及终产物替比培南酯的纯度和收率的方法(替比培南酯纯度达99.21%~99.78%,收率达88.7%~92.1%)[22],且发明了一种替比培南酯颗粒剂的制备方法[23]。成都倍特药业有限公司在2017年公开了一种替比培南杂质P8的合成方法[24]。
3.小结
综上所述,目前现有的制备替比培南侧链的合成路线均有一定限制,无法被广泛采用,也限制了替比培南抗生素的生产和推广。近年来,连续流微反应技术对于学术界和工业界的反应研究和优化变得越来越重要,连续流模式可通过在反应期间调整参数来轻松改变条件。连续流微反应系统可实现快速温度变化、低滞留量和快速优化周期。优化后,通过流系统的不同运行时间,可以很容易地将所需的反应产物量放大到一定程度。未来可应用该技术解决合成路线中危险系数高、难度高、收率低的部分操作以期促进替比培南抗生素的生产和应用。
