葛根素聚乳酸-羟基乙酸共聚物纳米粒制备及质量评价*
2022-06-29王璐璐甘昌冉韦馨琳胡婧怡秦贞苗
王璐璐,甘昌冉,韦馨琳,胡婧怡,朱 蓉,秦贞苗
(海南医学院药学院,海南 海口 571199)
滴眼液是治疗眼部疾病最方便、安全并且病人依从性最好的无创式眼用制剂[1]。但由于角膜屏障和泪液冲刷,滴眼给药后基本只有5%的药物能够到达内眼病灶部位[2],生物利用度低,需要通过频繁高浓度给药才能发挥治疗作用,但大量药物流失进入循环系统,易产生全身不良反应。因此寻求能提高药物生物利用度或使药物缓慢释放的眼部给药制剂尤为重要。纳米粒(Nanoparticles,NPs)是一种常用的纳米递药载体,能实现对难溶性药物的包载和输送,控制药物释放速度,延长药物在角膜的滞留时间,提高药物的眼部生物利用度[3-4]。
葛根素(Puerarin,Pue)是从豆科植物野葛PuerariathunbergianaBenth.根中提取的一种黄酮苷类化合物[5],具有扩张血管,降低眼压,改善微循环等作用[6]。目前市售的葛根素滴眼液主要用于青光眼的治疗,但是患者滴眼后由于不断眨眼活动导致药液大量流失,治疗效果显著降低[7]。本文以聚乳酸-羟基乙酸共聚物(polylactic-coglycolic acid,PLGA)为载体材料,结合单因素和正交设计试验考察Pue-PLGA NPs最优制备工艺,并对其进行初步的质量评价,以期为葛根素新型眼用制剂的开发奠定试验基础。
1 材料与方法
1.1 仪器与材料
UltiMate 3000型高效液相色谱仪,赛默飞世尔科技(中国)有限公司;Scientz-IID型超声波细胞粉碎仪,宁波新芝生物科技股份有限公司;Delsa Nano C型纳米粒度/ζ电位分析仪,美国Beckman Coulter公司;Micro 21R型台式冷冻离心机,赛默飞世尔科技(中国)有限公司;SN-MS-3D型三联磁力搅拌器,上海尚仪仪器设备有限公司;AX224ZH型电子天平,奥豪斯(常州)仪器有限公司;XW-80A型旋涡混合器,上海精科实业有限公司。
葛根素原料药(纯度≥98%),陕西林洲生物科技有限公司;葛根素对照品(纯度≥99%),中检所;1%葛根素滴眼液,浙江平湖莎普爱斯制药厂;PLGA(MW=20000,LA/GA=75/25),济南岱罡生物技术有限公司;聚乙烯醇(PVA,0588低粘度型),阿拉丁试剂(上海)有限公司;2-羟基-β-环糊精(2-HP-β-CD),阿拉丁试剂(上海)有限公司;超滤管(0.5 mL,3KD),密理博;甲醇为色谱纯,美国BCR公司;其他试剂均为分析纯。
1.2 Pue-PLGA NPs的制备
采用改良乳化溶剂挥发法[8]制备Pue-PLGA NPs。精密称取处方量葛根素和PLGA溶于适量乙醇-二氯甲烷(2:3)混合溶液中,形成有机相,另配制PVA和2-HP-β-CD混合溶液为水相,将有机相用注射器缓慢加入到水相中,冰浴下间断超声20 min(功率200 W,工作2 s,间隔2 s),形成初乳,继续磁力搅拌15 h,挥去有机溶剂,适量双蒸水定容,调节pH在7.0~7.5之间,即得Pue-PLGA NPs。同法制得不含Pue的空白PLGA NPs。
1.3 Pue-PLGA NPs中葛根素含量测定方法的建立
1.3.1 色谱条件
色谱柱:Thermo C18(5 μm,150 mm×4.6 mm);流动相:甲醇-水(25:75);检测波长:250 nm;流速:1.0 mL/min;柱温:30 ℃;进样量:10 μL。
1.3.2 溶液配制
对照品溶液:精密称取葛根素对照品10 mg置25 mL容量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得浓度为400 μg/mL的对照品溶液。
供试品溶液:精密量取Pue-PLGA NPs 1 mL置10 mL容量瓶中,加1 mL乙腈超声破乳,使用流动相稀释至刻度,摇匀,即为供试品溶液。
空白溶液:取适量PLGA NPs,同“供试品溶液”法制备空白溶液。
1.3.3 专属性试验
分别取对照品溶液、供试品溶液和空白溶液按“1.3.1”项下色谱条件进样测定,记录色谱图(图1),可见葛根素与基质成分分离情况良好,处方中的辅料对样品测定无干扰。

图1 高效液相色谱图Fig.1 HPLC chromatograms
1.3.4 线性关系
精密量取葛根素对照品溶液适量,分别配制成浓度为20、40、60、80、100、120和140 μg/mL的系列标准溶液,用0.22 μm滤膜过滤,取续滤液10 μL注入液相色谱仪,按“1.3.1”项下色谱条件测定。以葛根素浓度C为横坐标,峰面积A为纵坐标进行线性回归,得回归方程为A=0.731C-0.9245,r=0.9994。结果表明葛根素在20~120 μg/mL浓度范围内具有良好的线性关系(图2)。
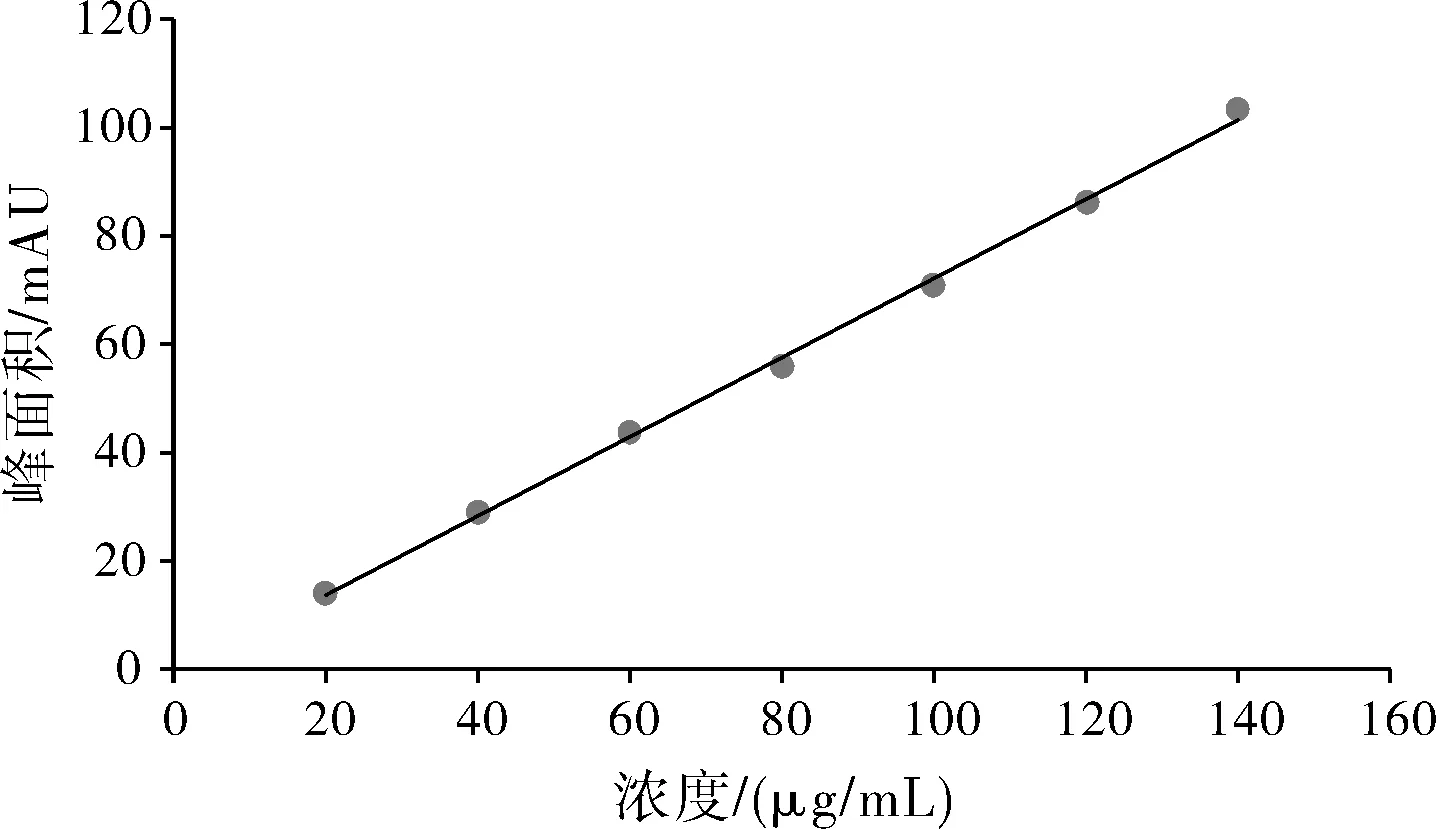
图2 葛根素对照品溶液标准曲线Fig.2 Calibration curve of puerarin reference solution
1.3.5 精密度试验
取“1.3.4”项下40 μg/mL的对照品溶液,连续进样6次,记录峰面积,计算RSD值为0.16%(n=6),表明仪器精密度良好。
1.3.6 重复性试验
精密量取同一批制备的Pue-PLGA NPs 1 mL,按“1.3.2”项下的供试品溶液操作,平行制备供试品溶液6份,在“1.3.1”项下色谱条件测定。结果测得Pue-PLGA NPs中葛根素的平均含量为987.6 μg·mL-1,RSD值为2.77%,表明方法重现性良好。
1.3.7 回收率试验
精密量取“1.3.2”项下的对照品溶液1.0 mL、1.25 mL、1.5 mL各3份置于10mL容量瓶中,分别加入空白PLGA纳米粒溶液0.5 mL,按“1.3.2”项下制备供试品溶液,在“1.3.1”项下色谱条件进样测定,测得平均回收率为95.72%,RSD值为1.93%(n=9)。
1.3.8 稳定性试验
取“1.3.6”项下供试品溶液,分别于0、1、2、4、8、12、24 h在“1.3.1”项下色谱条件测定,测得峰面积RSD值为1.78%,表明供试品溶液在室温24 h内稳定。
1.4 Pue-PLGA NPs包封率的测定
采用超滤离心法测定Pue-PLGA NPs的包封率。
精密量取“1.3.2”项下的对照品溶液1.0 mL、1.25 mL、1.5 mL各3份置于2 mL容量瓶中,分别加入空白PLGA纳米粒溶液0.5 mL,加水定容至刻度。精密量取0.4 mL置超滤管中,10000 rpm保持4 ℃离心30 min,精密量取接收管离心液体0.3 mL置1 mL容量瓶中,加流动相稀释定容,在“1.3.1”项下色谱条件进样测定葛根素含量,计算回收率。结果表明低、中、高不同含量的葛根素溶液加入到空白PLGA纳米粒溶液中后,回收率分别为98.38%、97.91%和101.46%,平均RDS值为1.27%,说明超滤法可有效分离纳米粒中的游离药物,可用于包封率的测定。
精密量取Pue-PLGA NPs 0.4 mL置超滤管中,10000 rpm保持4 ℃离心30 min,精密量取接收管离心液体0.3 mL置1 mL容量瓶中,加流动相稀释定容,在“1.3.1”项下色谱条件进样测定游离药物的含量M游离。按“1.3.2”项下制备供试品溶液过滤进样,测定Pue-PLGA NPs滴眼液中总的药物含量M总。按公式包封率EE=[(M总-M游离)/M总]×100%计算Pue-PLGA NPs的包封率。
1.5 Pue-PLGA NPs粒径的测定
取适量Pue-PLGA NPs用去离子水稀释10倍,用激光粒度测定仪测定Pue-PLGA NPs滴眼液的平均粒径和分布。
1.6 Pue-PLGA NPs处方工艺单因素考察
以粒径、包封率为评价指标,采用单因素实验考察油水比例、PVA浓度、PLGA浓度、药物浓度、超声时间、搅拌挥发时间等6个因素对Pue-PLGA NPs的影响(表1,实验中考察主要因素时其余因素全部选择水平3)。
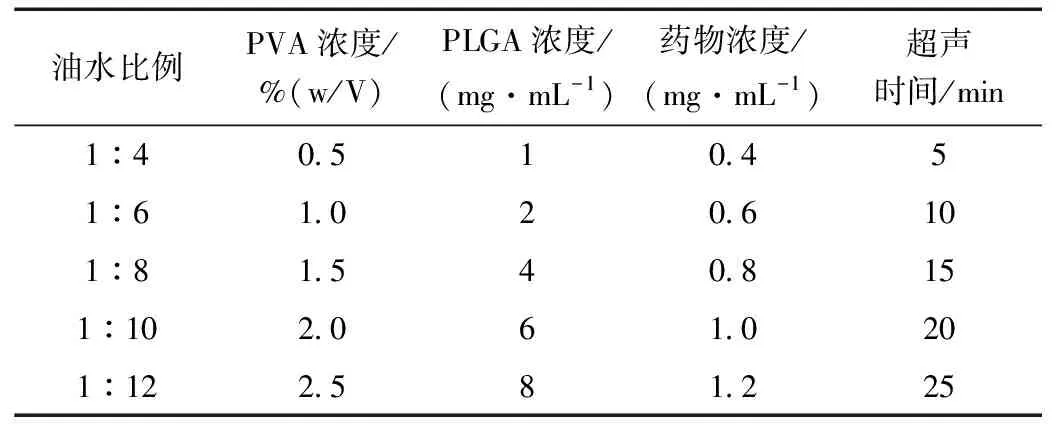
表1 单因素试验表Table 1 Single factor test table
1.7 Pue-PLGA NPs正交试验

表2 正交试验因素水平表Table 2 Orthogonal factor level table
在单因素实验的基础上,选取对Pue-PLGA NPs制备影响较大的3个因素:乳化剂浓度、PLGA浓度、超声时间,每个因素选择3个水平,按L9(34)正交表设计进行实验(表2)。以粒径和包封率为评价指标,采用综合加权评分法[9]对试验结果进行分析,粒径和包封率权重系数分别为0.4和0.6,按下面公式计算综合评分,并对试验结果进行直观分析和方差分析。

2 结果和讨论
2.1 处方工艺单因素条件对Pue-PLGA NPs的影响
2.1.1 油水比例的考察
固定水相体积不变,改变油相体积进行处方筛选,结果见图3。结果表明,随着油相用量的减小,纳米粒粒径先减小后增大,包封率先增大后减小。这是因为随着油相用量的减小,油相在水相中可以充分分散,形成粒径更小、更多的纳米粒[10];但是随着油相用量继续减小,一方面分散到水相的量减少后引起体系黏度增加,粒子聚集[11],另一方面油相溶解的药物和PLGA用量也减小,因此使得粒径增大,包封率下降。
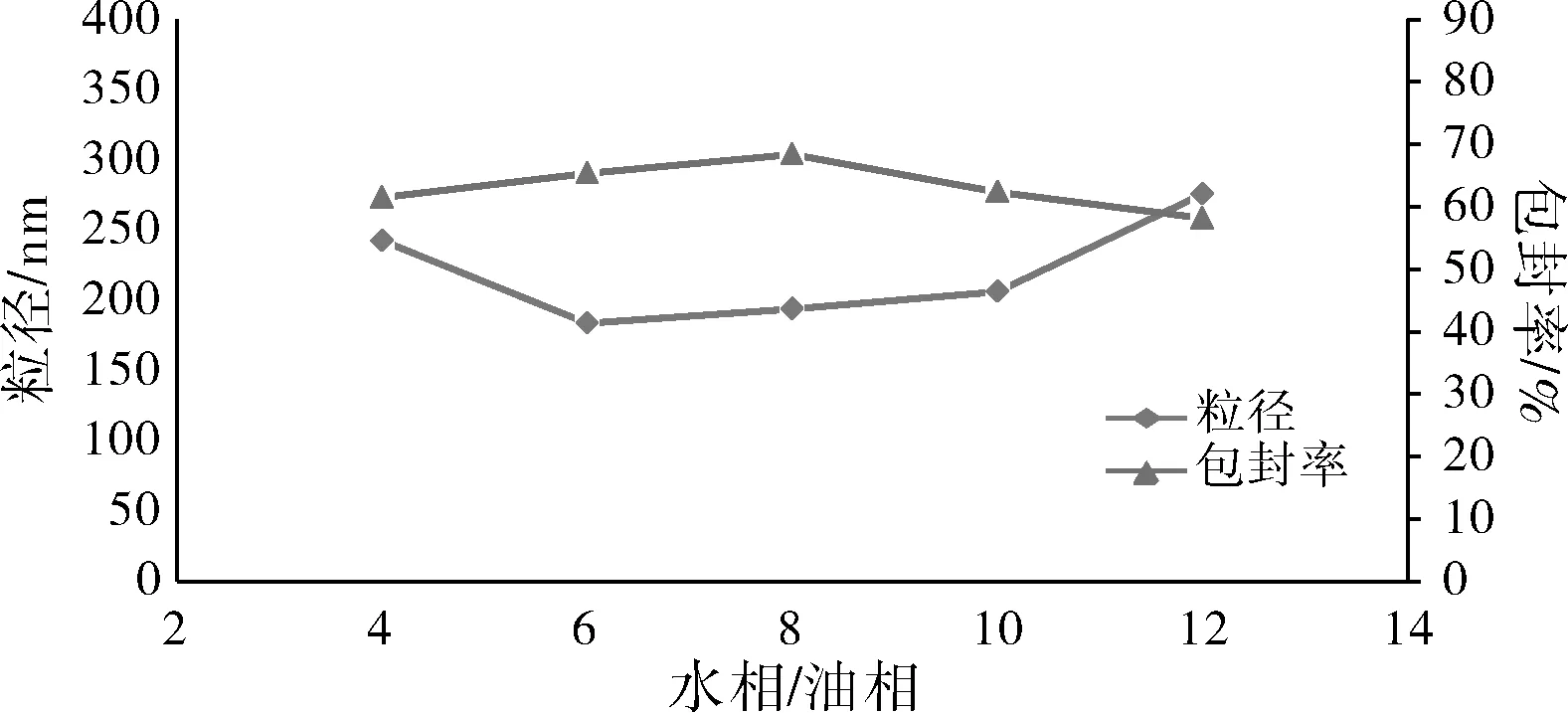
图3 油水比例对纳米粒粒径和包封率的影响Fig.3 Effect of oil phase/aqueous phase volume ratio on nanoparticle size and entrapment efficiency
2.1.2 PVA浓度的考察
分别采用0.5、1.0、1.5、2.0、2.5%不同质量浓度的PVA溶液进行处方筛选,结果见图4。结果表明,随着PVA质量浓度的增大,纳米粒的粒径逐渐增大,包封率变化不大。这是因为乳化剂用量增大时,体系的黏度增加[12],粒子间发生黏连聚集,形成的颗粒逐渐增大。

图4 PVA浓度对纳米粒粒径和包封率的影响Fig.4 Effect of PVA concentration on nanoparticle size and entrapment efficiency
2.1.3 PLGA浓度的考察
分别采用1、2、4、6、8 mg·mL-1不同质量浓度的PLGA进行处方筛选,结果见图5。结果表明,随着PLGA质量浓度的增大,纳米粒粒径和包封率逐渐增大。这是因为PLGA用量增大后,有机相的黏度增加,不易在水相中分散均匀[10],形成的纳米粒粒径逐渐增大;同时,PLGA用量增大,包封的药物增加,包封率也逐渐增加。

图5 PLGA浓度对纳米粒粒径和包封率的影响Fig.5 Effect of PLGA concentration on nanoparticle size and entrapment efficiency
2.1.4 药物浓度的考察
分别采用0.4、0.6、0.8、1.0、1.2 mg·mL-1不同质量浓度的药物进行处方筛选,结果见图6。结果表明,随着药物质量浓度的增大,纳米粒粒径未见明显变化,包封率先增大后减小。这是因为药物浓度较少时,乳化剂的增溶作用使部分药物溶解到水相中,包封率较小。随着药物浓度的增加,乳化剂增溶作用减弱,包封率逐渐增加。但当药物浓度过大时,在PLGA不变的情况下,导致药物包封不完全,包封率下降。
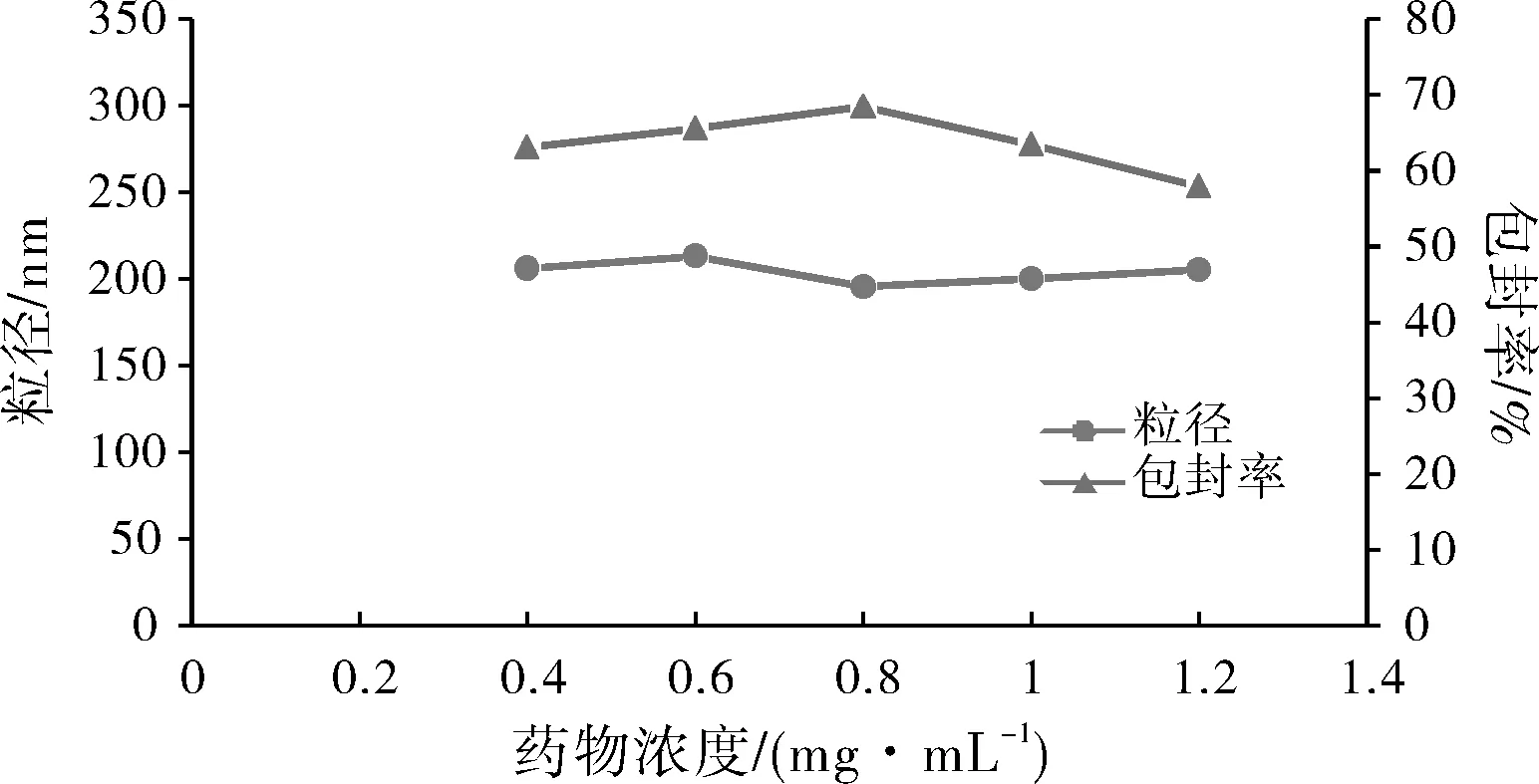
图6 药物浓度对纳米粒粒径和包封率的影响Fig.6 Effect of drugconcentration on nanoparticle size and entrapment efficiency
2.1.5 超声时间的考察
分别采用5、10、15、20、25 min不同超声时间进行处方筛选,结果见图7。结果表明,随着超声时间的增加,纳米粒粒径逐渐减小,包封率先增大后减小。这是因为延长超声时间,有机相在水相中能充分乳化分散,生成的纳米粒较多、粒径较小。但当超声时间过长时,温度升高,影响药物和体系的稳定性。
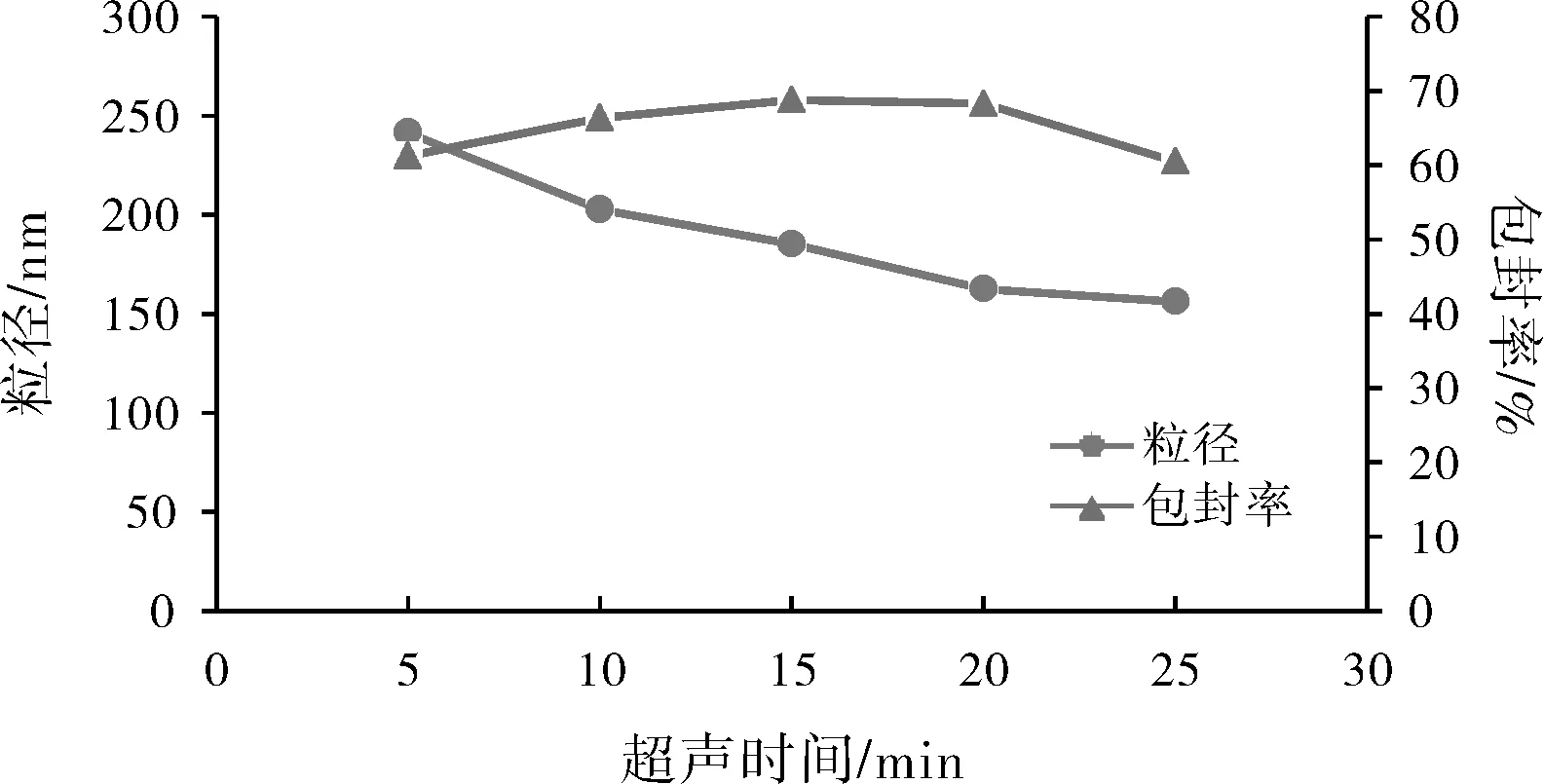
图7 药物浓度对纳米粒粒径和包封率的影响Fig.7 Effect of ultrasonic time on nanoparticle size and entrapment efficiency
2.2 正交试验设计对处方及制备工艺的优化
在单因素实验的基础上,选取对Pue-PLGA NPs制备影响较大的3个因素:乳化剂浓度、PLGA浓度、超声时间,每个因素选择3个水平制备9批纳米粒。从直观分析结果(表3)可知,影响Pue-PLGA NPs制备的因素主次顺序为C(超声时间)>A(PVA浓度)>C(PLGA浓度),最优处方为A1B2C3,即PVA浓度为1.0%,PLGA浓度为4 mg·mL-1,超声时间为20 min;从方差分析结果(表4)可知,PVA浓度和超声时间对纳米粒的制备影响显著,PLGA浓度对纳米粒制备无显著性影响。
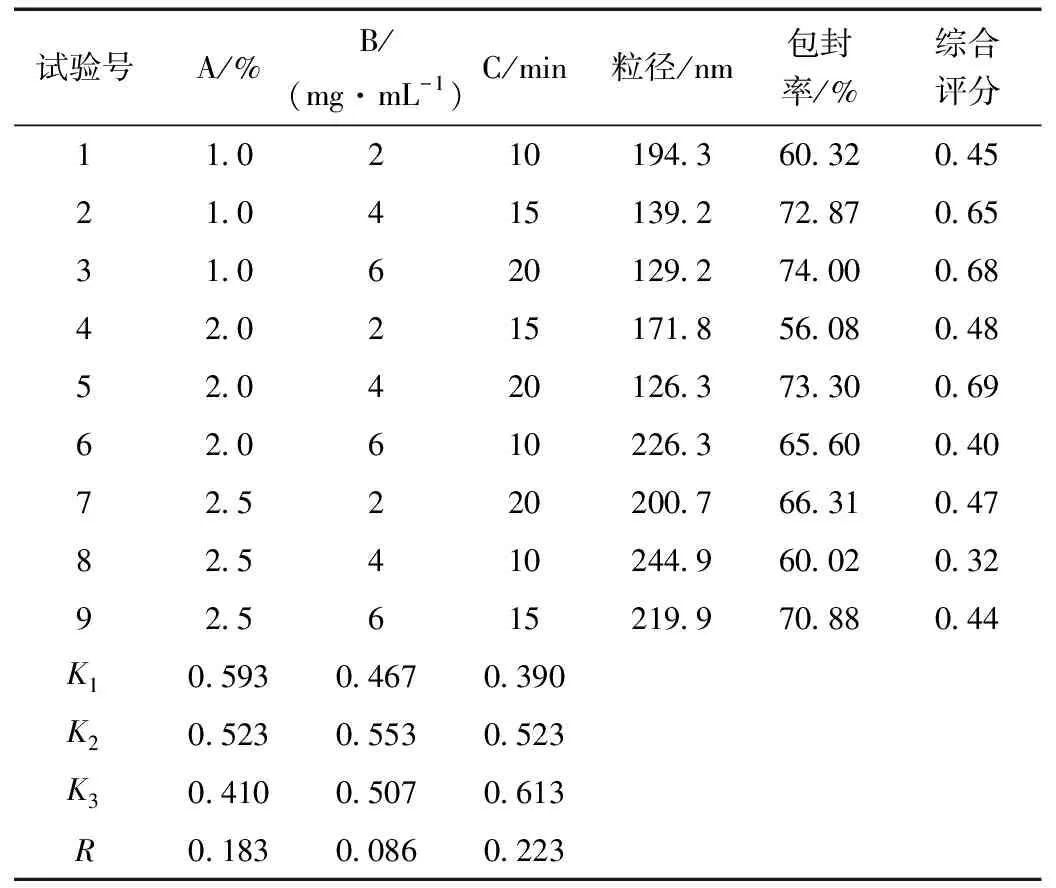
表3 正交实验设计和结果Table 3 Orthogonal experimental design and results
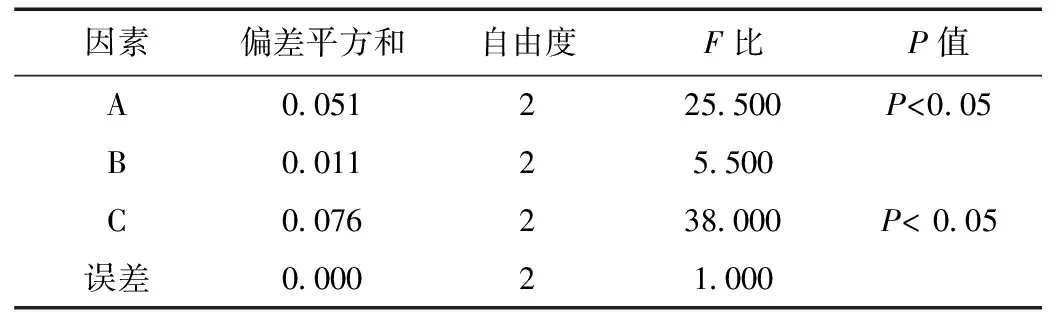
表4 方差分析Table 4 Analysis of variance
2.3 Pue-PLGA NPs处方工艺验证
按最优处方制备3批Pue-PLGA NPs,测得平均粒径为(141.4±2.4)nm,平均包封率为(71.31±0.57)%,批间重复性好,处方工艺稳定。
2.4 Pue-PLGA NPs的表征
2.4.1 形态观察

图8 Pue-PLGA NPs透射电镜图Fig.8 Transmission electron microscopic image of Pue-PLGA NPs
取少量Pue-PLGA NPs加水稀释后,置于铜网上,固定10 min后用滤纸从铜网边缘吸干多余的溶液,室温过夜自然晾干,置于透射电镜下观察其形态,结果见图8。由图8中可见制得的Pue-PLGA NPs呈球型,大小及分布均匀,粒子间无明显黏连。
2.4.2 粒径分布及Zeta电位
取少量Pue-PLGA NPs加水稀释后,于激光粒度测定仪测定其粒径分布和Zeta电位,结果见图9和图10。图9和图10可见纳米粒粒径呈分布均匀,平均粒径为(149.2±4.8)nm,PDI 为0.111±0.05,Zeta电位为-(5.73±0.12)mV。表明Pue-PLGA NPs体系较稳定,粒子不易发生聚集,与透射电镜观察结果一致。

图9 Pue-PLGA NPs粒径分布Fig.9 Particle size distribution of Pue-PLGA NPs

图10 Pue-PLGA NPs Zeta电位Fig.10 Zeta potential of Pue-PLGA NPs
3 结 论
本文以PLGA为载体材料,采用改良的乳化溶剂挥发法制备Pue-PLGA NPs。PLGA是由丙交酯LA和乙交酯GA开环无规共聚合得到的,作为药物载体材料具有生物可降解的优点,同时能控制药物的释放速率,显著提高药物的生物利用度[13]。在实验中发现当使用乙醇:二氯甲烷有机溶剂混合液的比例为3:2时,PLGA不能完全溶解,当改为乙醇:二氯甲烷(2:3)时,PLGA则可以完全溶解,而且酯封端的PLGA比酸封端的PLGA测得的包封率高,说明乙醇对PLGA的溶解度不高,Pue是脂溶性药物,更容易和酯封端的PLGA产生吸附作用。
粒径和包封率是载药纳米粒制备工艺的2个重要评价指标。研究发现,微粒粒径小于200 nm时,能增加药物透过角膜的量,药物多分布于视网膜;但微粒粒径小于20 nm时,只有极少量到达视网膜,这主要原因是微粒过小时易在眼周循环过程中清除[14-15]。另外,粒径大小是影响纳米制剂释放的主要因素,当粒径为纳米范围时能延长药物释放的时间,达到缓释的效果。本实验考察了低速离心法、透析法和超滤法测定纳米粒包封率。结果发现,超滤法能有效分离纳米粒和游离药物,测得的包封率数值差异小,回收率高;对于低速离心法,在5000 r·min-1条件下离心30 min,上清液透镜结果显示仍有游离药物晶体存在,而且包封率测定结果平行性不好;对于透析法,在100 r·min-1,37 ℃气浴恒温振荡,随着振荡时间的增加,PLGA容易降解,导致纳米粒发生药物泄露,测得的包封率偏低。
本研究初步优化了Pue-PLGA NPs的处方工艺,制得的纳米粒粒径适宜、分布均匀,包封率较高,形态呈球形,体系稳定,为进一步考察其角膜渗透性和药动学特征提供实验基础。
