面包面条优质小麦转录组学分析
2021-06-09巨伟王红日薛春芝庞亚男吴德豪郭宪峰程敦公韩冉刘爱峰李豪圣刘建军曹新有宋健民訾妍
巨伟,王红日,薛春芝,庞亚男,吴德豪,郭宪峰,程敦公,韩冉,刘爱峰,李豪圣,刘建军,曹新有,宋健民,訾妍
(1.山东省农业科学院作物研究所,山东 济南 250100;2.山东鲁研农业良种有限公司,山东 济南 250100;3.菏泽市牡丹区种子资源开发保护中心,山东 菏泽 274000)
转录组测序是解析生物经济性状形成分子机制、发掘基因资源的重要手段之一[1],该技术为差异基因表达分析、功能基因和转录因子挖掘、等位基因特异性表达研究等提供了新的手段,尤其是对全面解析复杂性状的遗传调控网络非常有效,已成功应用于水稻、玉米、小麦、黄瓜、黄豆、番茄等多种作物研究。因此,获得高质量的转录组对于作为重要粮食作物之一的小麦具有重要的研究意义。近年来,针对小麦抗旱、耐热、抗病等性状进行了一些转录组研究,筛选出部分目的基因,并对筛选到的基因进行了深入分析[2-5]。
小麦食品加工品质是决定其生产应用和市场竞争力的关键因素,小麦品质主要受蛋白、淀粉等籽粒主要成分理化特性的控制,而蛋白和淀粉的合成受一系列基因和调控因子的复杂网络调控,因此利用RNA-Seq技术构建不同面包、面条品质类型典型品种籽粒转录组文库,筛选差异表达基因,发掘调控面包、面条品质的关键基因和调控因子,解析小麦品质形成的生理代谢机制和调控网络,对于指导我国小麦品质遗传改良具有重要意义。小麦品质性状非常复杂,过去研究多局限于单一或个别性状的影响和调控分析,研究方法也多是统计分析,很少在全基因组水平全面解析小麦品质的遗传调控网络。RNA-Seq技术的发展,为从转录组水平全面解析小麦品质遗传调控提供了可能。Yu等[6]通过动态转录组分析,鉴定出8个对淀粉与蛋白质合成以及压力胁迫响应相关的基因。利用我们前期研究中筛选出的6类有代表性的小麦材料:面包优质小麦、面包劣质小麦、面条优质小麦、面条劣质小麦、面包和面条兼优小麦、面包和面条均劣小麦各2份,本研究利用RNA-Seq技术分析了其灌浆不同时期(开花后7、17、27 d)的基因表达情况,以期筛选调控面包、面条品质性状的差异基因和调控因子,结合蛋白和淀粉理化特性筛选关键基因进行qRT-PCR验证,这对全面解析小麦品质遗传调控网络、促进小麦品质遗传改良具有重要意义。
1 材料与方法
1.1 试验材料与种植方法
供试品种为6类12个小麦品种,分别是面包优质小麦(GB)JM4072、ZM12,面条优质小麦(GN)JM19、ZM18,面包劣质小麦(BB)TN18、QF1,面条劣质小麦(BN)LM1、LM19,面包面条均优小麦(GBN)JM20、ZM366,面包面条均劣小麦(BBN)LM14、XM18。
试验于2014—2016年在山东省农业科学院实验站(36°42′N、117°05′E)进行。试验采用随机区组设计,播种密度为3×106株/hm2,行距25 cm,小区面积6 m2(4.0 m×1.5 m),重复3次。2014年10月和2015年10月播种,2015年6月和2016年6月收获,均按高产田常规栽培措施管理。
1.2 试验方法
1.2.1 样本采集及RNA提取、纯化 分别于花后7、17、27 d取同一天开花的小穗中部籽粒,送至广州基迪奥生物科技有限公司,利用Trizol法提取总RNA,采用DNaseⅠ(RNase-free)消化所得RNA样本中的残余DNA。
1.2.2 转录组测序 样品提取总RNA后,经富集、打断、六碱基随机引物(random hexamers)合成、末端修复、加碱基A、加测序接头、PCR扩增等过程制备测序文库,构建好的文库用Illumina-HiSeqTM进行测序。
1.2.3 RNA-Seq质量评估和序列比对 在全部测序结果中选择错误率小于1%的用于后续转录组分析,更严格过滤得到high quality clean reads,同时计算Q20和Q30。小麦参考基因组从http://www.wheatgenome.org/下载,选取HISAT软件将过滤后的测序序列进行基因组定位分析。样本之间(包括3个生物学重复)的基因表达水平则采用皮尔逊相关性检测进行分析。
1.2.4 差异基因筛选 将同一类型的两个品种作为一组,进行不同组间小麦品种的差异基因分析。基因表达量的计算采用FPKM(fragments per kilobase of exon per million fragments mapped reads)法[7]。使用DESeq R软件包(1.18.0)[8]对不同类型小麦之间的差异表达基因进行研究。当FDR校正后的p值<0.05且时则认为该基因表达差异显著。
1.2.5 GO注释和KEGG分析 Gene Ontology(简称GO,http://www.geneontology.org/)富集分析采用软件为GOseq[9],GO分为分子功能(molecular Function)、生物过程(biological process)和细胞组成(cellular component)三个部分。
KEGG(Kyoto Encyclopedia of Genes and Genomes,http://www.kegg.jp/)是系统分析基因功能、基因组信息数据库,分析内容包括碳水化合物、核苷、氨基酸等的代谢及有机物的生物降解[10]。
1.2.6 qRT-PCR验证 为了验证RNA-Seq检测出的基因表达水平是否正确,从RNA-Seq测序用总RNA中取出1μg用于qRT-PCR实验。使用Primer Premier 5软件进行引物设计,引物序列由擎科生物技术有限公司合成,釆用PAGE纯化(表1)。
使用罗氏公司的LightCycler 480Ⅱ进行荧光定量PCR。荧光定量PCR反应体系10μL包括:2×SYBR Premix Ex TaqⅠ5μL,正向引物与反向引物各0.5μL,cDNA 1μL,ddH2O 3μL。PCR程序为:95℃1 min预变性;40×(95℃15 s,55℃15 s,72℃15 s)收集荧光信号;95℃1 s,60℃1 s,95℃1 s,37℃1 s为熔解曲线阶段。采用2-ΔΔCt法[11]分析目的基因的相对表达量。
2 结果与分析
2.1 测序结果质量评估
对原始数据进行数据过滤去除大量的测序接头序列、低质量的读段、序列N的比例大于10%的reads,最终得到2 882 357 526条clean reads,每个样本测序量超过10 G,达到文库采样深度的要求。质量达到Q30级别的碱基占总碱基数的84%以上(表2)。51%以上的reads可以比对到参考基因组上,50%以上的reads能够比对到唯一转录本上,2%左右的reads可比对到多个转录本上。且reads打断随机性好,在参考基因组各个位置分布均匀。比对到唯一转录本的reads可以用来进行下一步的分析工作。
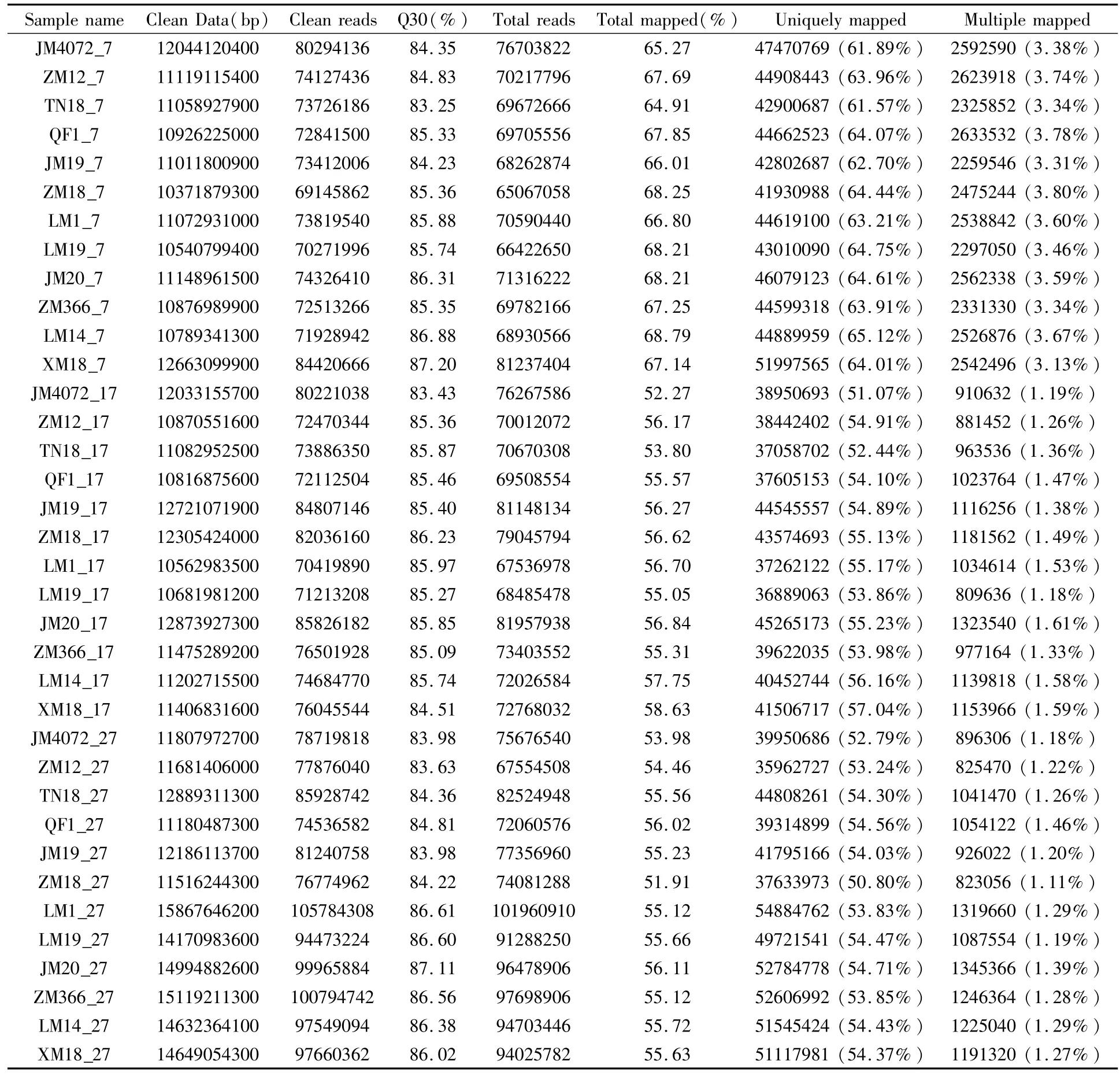
表2 RNA-Seq测序结果质量评估
2.2 不同类型面包、面条小麦不同时期差异表达基因的分析
为了鉴别6类不同加工品质小麦的差异表达基因(DEGs),对组间基因表达量进行了比对分析。以同期GB小麦为对照,花后7 d共找到4 529个差异表达基因,其中有2 549、1 980个基因表达量分别呈上调和下调;花后17 d共找到3 198个差异表达基因,其中有1 775、1 423个基因表达量分别呈上调和下调;花后27 d共找到2 955个差异表达基因,其中,1 731、1 224个基因表达量分别呈上调和下调(图1)。6类品种在花后7 d差异表达基因最多,其次是花后17 d,花后27 d最少,这可能是因为灌浆前期籽粒膨大生长阶段差异基因较多。
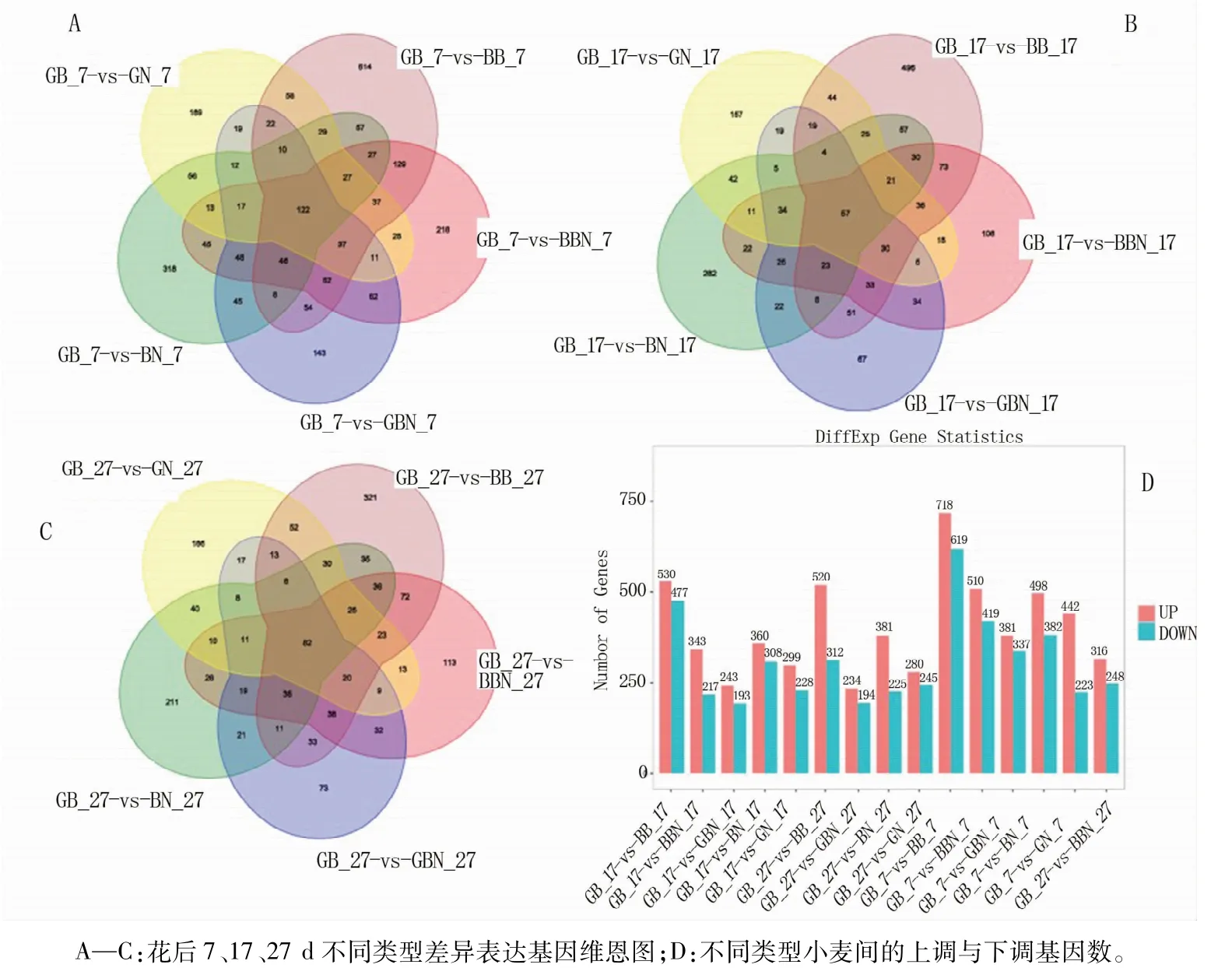
图1 不同类型小麦组间差异表达基因的维恩图
2.3 差异表达基因的筛选及GO注释和KEGG分析
发现花后7、17、27 d分别有1 743、1 195、981个DEGs在优质小麦(GN与GBN)中上调,在劣质小麦(BB、BN与BBN)中下调,对差异表达的基因进行GO功能分类分析发现,三个时期差异基因均主要分布在生物过程(biological process)的metabolic process和cellular process、细胞成分(cell component)的cell和cell part以及分子功能(molecular function)的binding和catalytic activity中。
利用KEGG分析研究了与品质相关的生物代谢通路。发现花后7 d筛选出的1 743个DEGs被富集到103条通路中,包括DNA及RNA的合成及转运、碳循环、氮代谢、氨基酸代谢、淀粉及糖代谢等,富集DEGs数目最多的代谢通路有:protein processing in endoplasmic reticulum(36)、starch and sucrose metabolism(22)、spliceosome(20)、plant-pathogen interaction(18)、RNA transport(17)(图2A)。花后17 d筛选出的1 195个DEGs被富集到98条通路中,包括DNA及RNA的合成及转运、植物激素信号转导、氨基酸的生物合成、碳循环等,富集DEGs数目最多的代谢通路有:RNA transport(15)、spliceosome(12)、biosynthesis of amino acids(12)、plant-pathogen interaction(12)(图2B)。花后27 d筛选出的981个DEGs被富集到89条通路中,包括DNA及RNA的合成及转运、氨基酸生物合成、内质网蛋白合成、淀粉及糖代谢等,富集DEGs数目最多的代谢通路有:RNA transport(14)、protein processing in endoplasmic reticulum(10)、spliceosome(9)、biosynthesis of amino acids(9)、starch and sucrose metabolism(9)(图2C)。
2.4 蔗糖与淀粉代谢通路有关的差异基因分析
对蔗糖与淀粉代谢通路进行研究发现,在以GB为对照的条件下,花后7、17 d及27 d BB与GB、BBN与GB间淀粉合成通路中差异表达基因最多。花后7 d,相较于GB小麦,其他类型小麦下调差异基因较多,花后17 d与花后27 d,BB小麦与GN小麦均为上调基因较多,其他类型为下调基因较多(表3)。
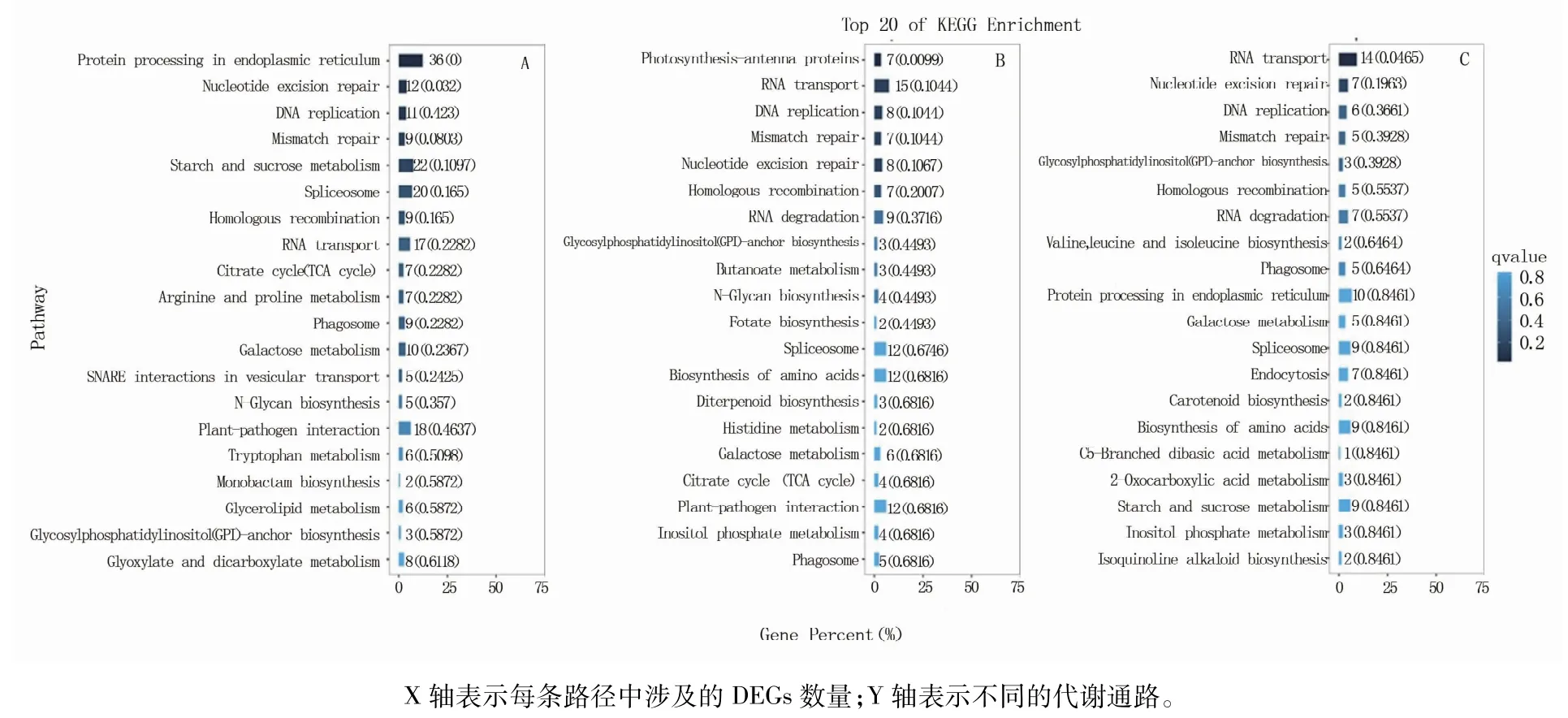
图2 花后7 d(A)、17 d(B)及27 d(C)差异表达基因KEGG富集分析
对淀粉与蔗糖代谢通路的差异表达基因进行分析,以GB小麦为对照,从花后7 d的BB、GN、BN、GBN、BBN品种中分别筛选到14、5、8、6和12个差异表达基因,从花后17 d的BB、GN、BN、GBN、BBN品种中分别筛选到14、4、6、4和4个差异表达基因,从花后27 d的BB、GN、BN、GBN、BBN品种中分别筛选到7、4、7、4和5个差异表达基因,主要涉及糖代谢过程中的蔗糖合成酶(sucrose synthase,SuS,EC2.4.1.13)和转化酶(invertase,INV,EC3.2.1.26)、蔗糖磷酸酯酶(sucrose phosphatase,SPP)、蔗糖磷酸合酶(sucrosephosphate synthase,SPS),淀粉代谢过程中的去分支酶(debranching enzyme,DBE)、α-淀粉酶(alpha-amylase,EC3.2.1.1)和 β-淀粉酶(beta-amylase,EC3.2.1.2)。
SPS基因花后7 d在BN小麦中上调,花后17 d在BB小麦中下调,花后27 d在GN、BN与BBN小麦中均上调。相反,SuS在花后7 d的BB、BN小麦中上调,在GN与BBN中下调,花后17 d的BB、GN、BN以及BBN小麦中均上调,花后27 d的BB小麦中上调;INV在花后7 d的BB、BN与GBN中均下调,花后17 d在BB、BN和GBN小麦中均下调,花后27 d在BB、GN、GBN、BBN小麦中均下调(表4—表6)。推测花后7 d GN小麦、GBN及BBN小麦蔗糖分解能力较弱;花后17 d GN小麦及BBN小麦蔗糖分解能力高于GB小麦;花后27 d GN、BN与BBN小麦的蔗糖合成能力高于GB小麦。
6-磷酸葡萄糖异构酶(glucose-6-phosphate isomerase,GPI,EC5.3.1.9)基因在花后7、17、27 d的BB小麦中均上调。DBE基因在花后不同时期其他5类小麦中均上调,推测与成熟期GB小麦总淀粉含量最低一致。α-淀粉酶和β-淀粉酶基因在花后7 d的BB小麦中下调,但花后17 d BB小麦中的α-淀粉酶上调,可以进一步推测是成熟期支链淀粉含量低于GB小麦的原因之一。
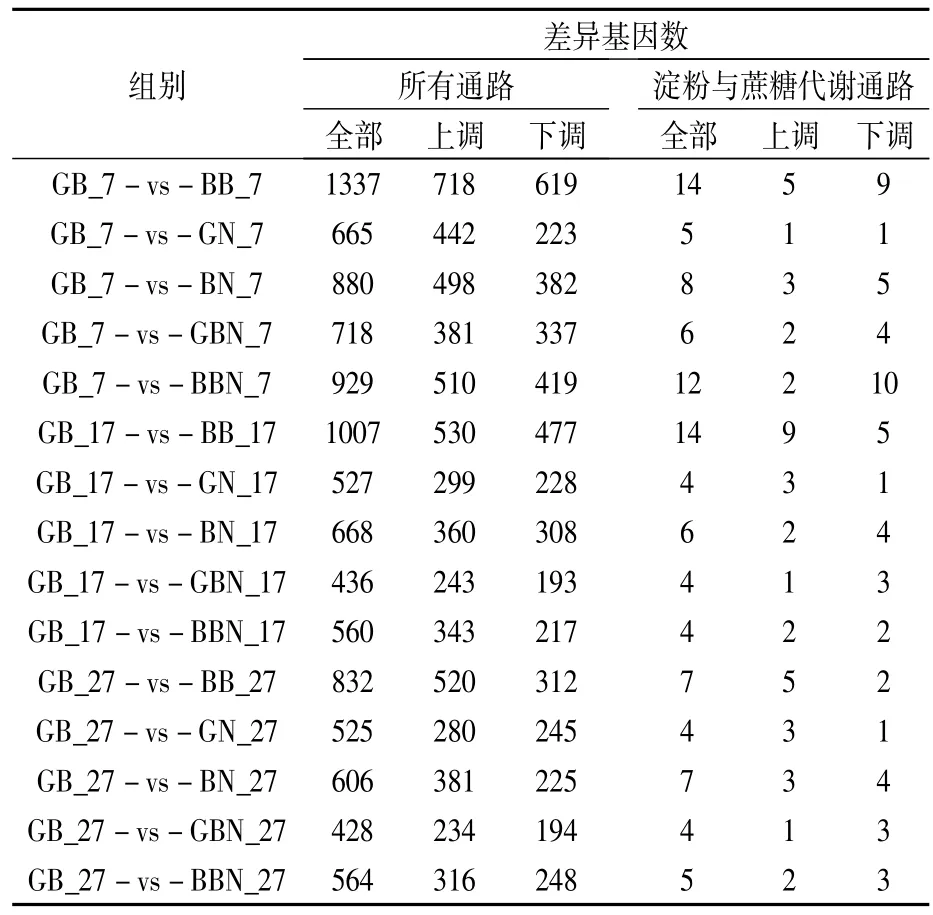
表3 不同时期不同类型DEGs及与淀粉合成相关的DEGs汇总
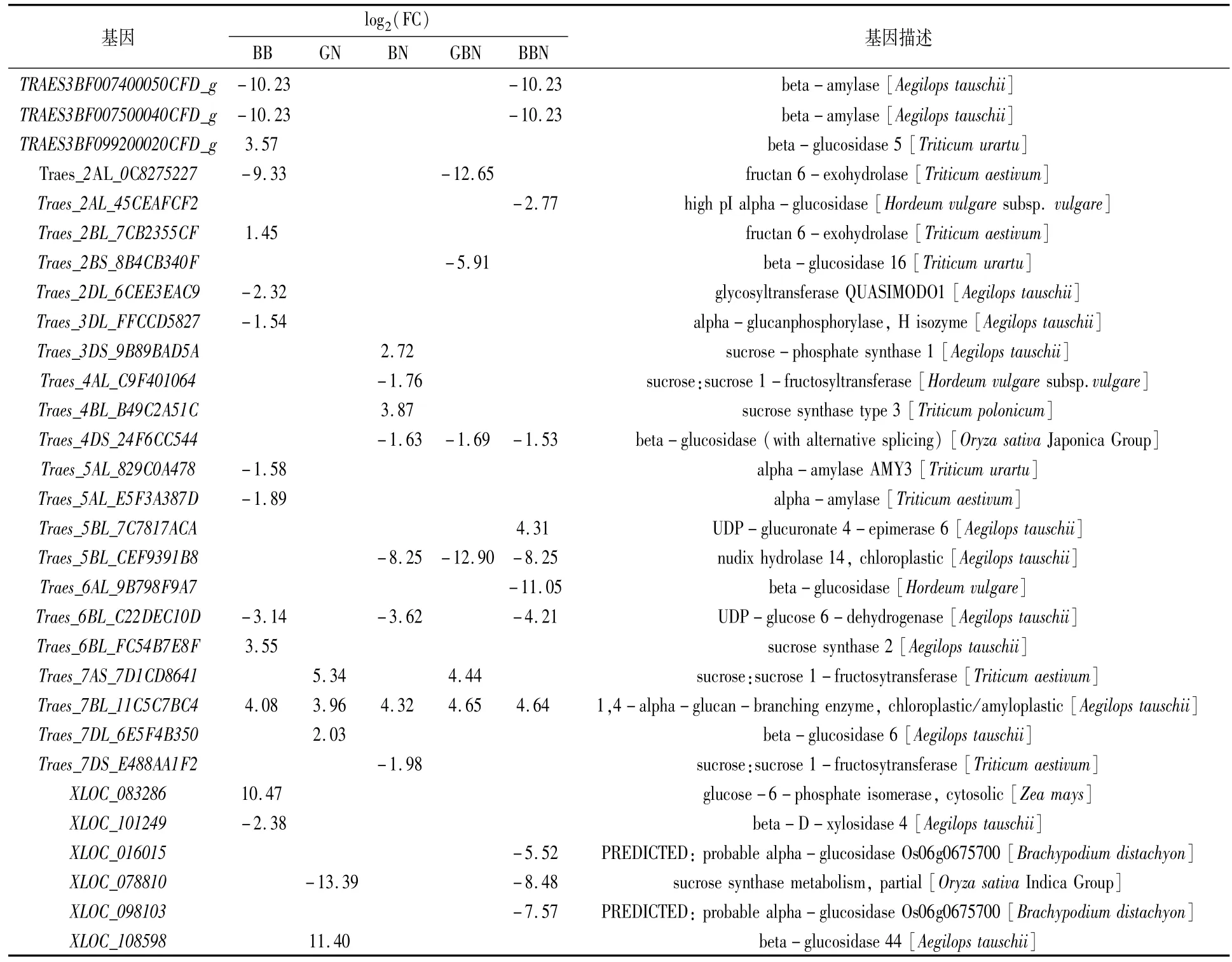
表4 花后7 d与蔗糖和淀粉代谢通路相关的DEGs
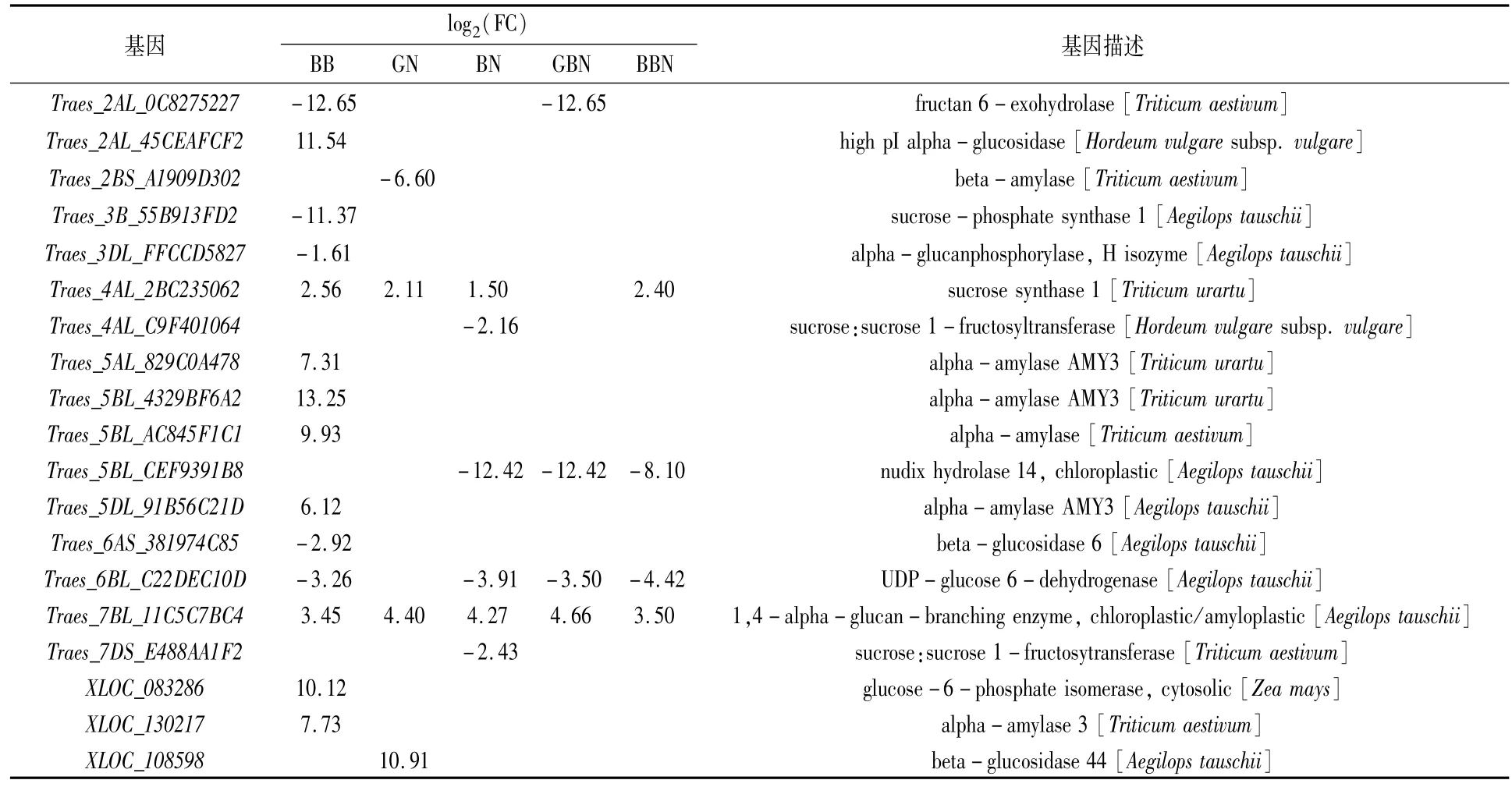
表5 花后17 d与蔗糖和淀粉代谢通路相关的DEGs
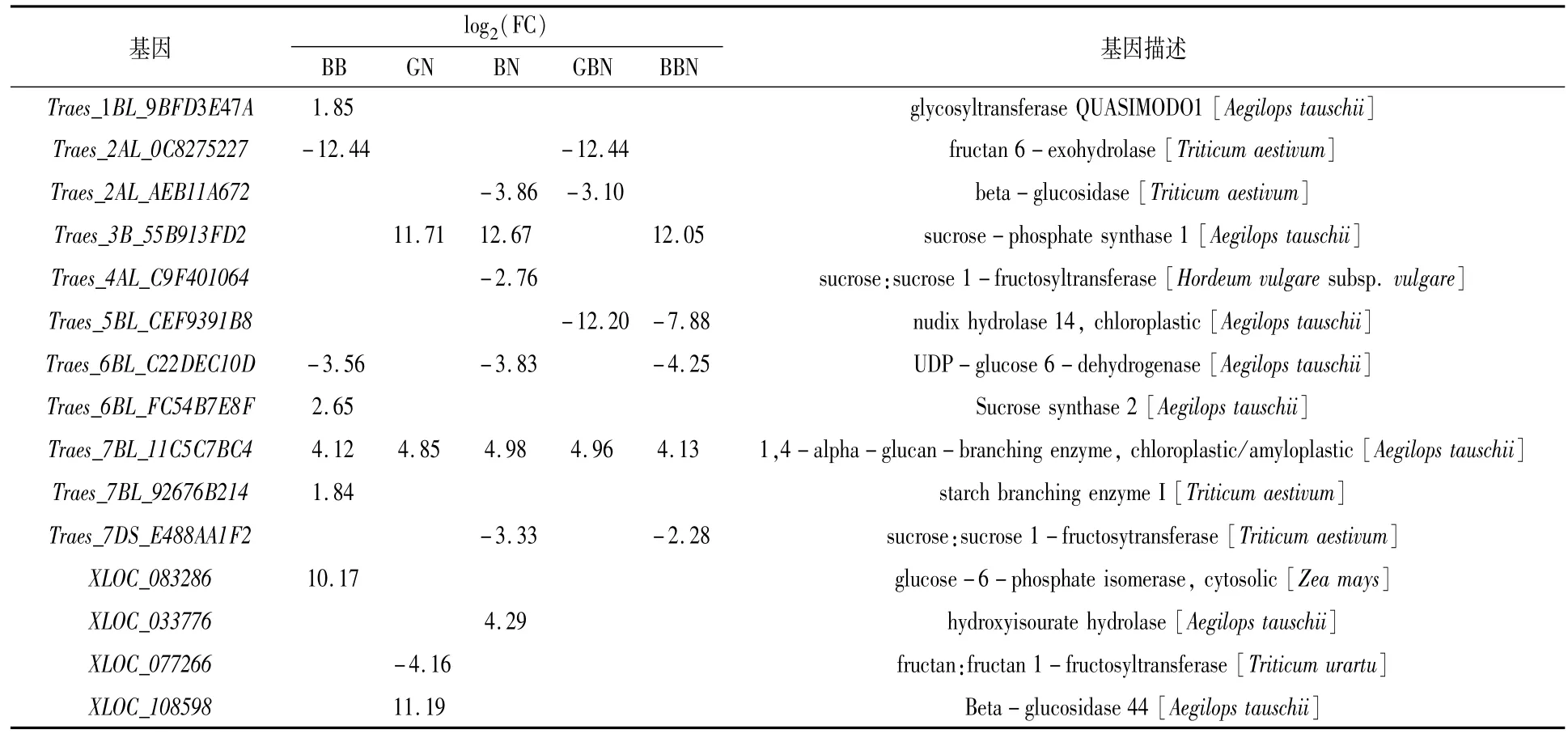
表6 花后27 d与蔗糖和淀粉代谢通路相关的DEGs
2.5 qRT-PCR验证差异表达基因
为验证转录组测序结果的真实性,从淀粉合成通路中随机挑选了7个候选基因(Traes_7BL_11C5C7BC4、Traes_7AS_7D1CD8641、Traes_3B_55B913FD2、Traes_6BL_C22DEC10D、Traes_2AL_0C8275227、Traes_6BL_FC54B7E8F和Traes_7DS_E488AA1F2)进行qRT-PCR验证(图3、4、5)。将验证结果与RNA-seq数据进行相关性分析,两者的相关系数较高且表达趋势基本相同,表明RNA-Seq的分析结果可靠。
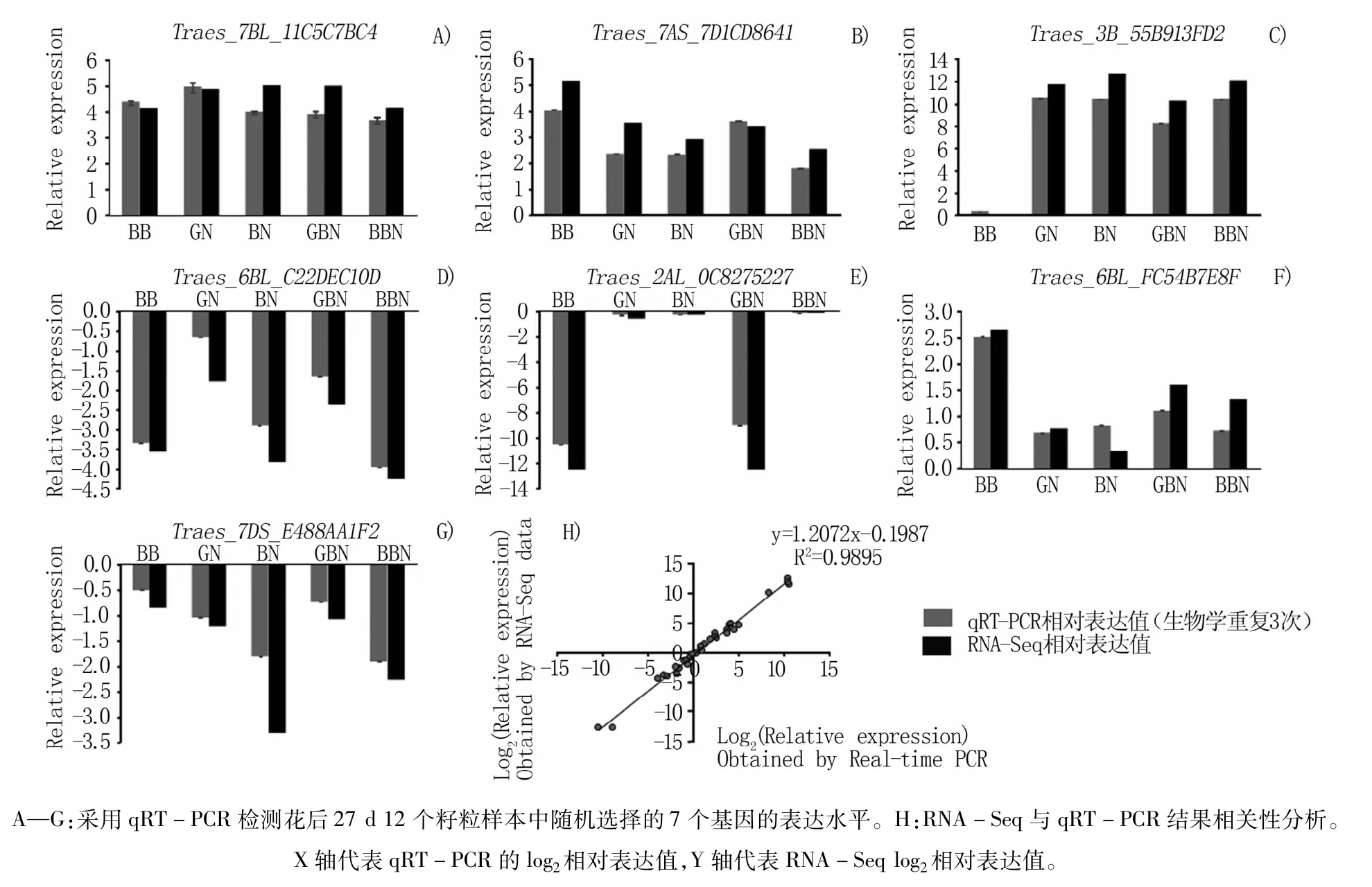
图5 qRT-PCR验证花后27 d RNA-Seq结果
3 讨论与结论
本研究采用RNA-Seq技术分析不同面包、面条品质类型小麦在花后7、17、27 d的差异表达基因。以相同时期面包优质小麦为对照,分别发现4 529、3 198、2 955个差异表达基因。通过对不同加工品质小麦差异表达基因的GO富集分析,发现不同类型小麦在淀粉代谢通路中的差异表达基因及其参与的生物过程以及本身的分子功能。与面包优质小麦进行比较,不同类型小麦在花后不同时期差异表达的基因主要参与代谢过程、细胞生理过程、细胞、细胞组分、结合功能以及催化活性。对不同类型小麦的KEGG通路注释结果发现,不同类型小麦之间的差异表达基因在蛋白质合成的内质网加工过程通路、淀粉和蔗糖代谢通路、剪接体通路、RNA转录通路、氨基酸合成通路以及核糖体通路等通路富集较多。
淀粉与蔗糖代谢在植物发育、抗逆反应等多个生物学过程中发挥重要作用。小麦淀粉含量及特性对小麦品质特别是面条品质影响较大,培育面包面条兼优小麦的其中一个思路即为在面包优质小麦的基础上提高面条品质。因此,我们重点对淀粉与蔗糖代谢通路进行分析。对淀粉与蔗糖代谢通路的差异表达基因进行分析,以面包优质小麦为对照,从花后7、17、27 d的面包劣质、面条优质、面条劣质、包条兼优以及包条均劣品种分别筛选到一些差异表达基因,主要涉及糖代谢过程中的蔗糖合成酶(SuS)和转化酶(INV)、蔗糖磷酸酯酶(SPP)、蔗糖磷酸合酶(SPS),淀粉代谢过程中的去分支酶(DBE)、α-淀粉酶和β-淀粉酶。在蔗糖代谢方面,蔗糖在蔗糖合酶和转化酶调控下参与淀粉的合成[12],蔗糖磷酸酯酶和蔗糖磷酸合酶在蔗糖合成中起关键作用[13,14]。INV对蔗糖的水解作用是单向的,而SuS能可逆地催化蔗糖代谢,通常认为SuS主要起水解蔗糖的作用。SPS在碳水化合作物代谢方面发挥重要作用,可以调控碳在淀粉合成和碳水化合物积累间的分配[15]。
催化6-磷酸果糖和6-磷酸葡萄糖相互转化的6-磷酸葡萄糖异构酶(GPI)[16]基因在花后7、17、27 d的BB小麦中均上调,而6-磷酸葡萄糖是合成ADPG的重要物质基础[17],因此推测BB籽粒中ADPG的合成能力高于GB小麦,这与BB小麦具有最高的总淀粉及直链淀粉含量相符合。α-淀粉酶与β-淀粉酶是淀粉水解过程中的关键酶,其中α-淀粉酶可以水解淀粉内部的α-1,4-糖苷键,β-淀粉酶能将直链淀粉分解为麦芽糖;淀粉脱支酶(DBE)专一水解α-1,6-糖苷键,支链淀粉经淀粉酶水解产生的极限糊精,由脱支酶水解去除α-1,6-糖苷键,再在α-淀粉酶和β-淀粉酶的作用下彻底水解[18]。
不同品质类型小麦籽粒中蔗糖代谢形成淀粉合成前体的相关酶类(SPS、INV、SuS)以及淀粉代谢相关酶类(DBE、α-淀粉酶与β-淀粉酶)存在差异,是总淀粉含量及其组分的差异原因之一。面包劣质小麦籽粒淀粉含量及直链淀粉含量均高于面包优质小麦,可能是由于其6-磷酸葡萄糖异构酶基因的高表达导致。其他5类小麦品种中DBE与α-淀粉酶基因均高于面包优质小麦,可能是面包优质小麦总淀粉含量较低、支链淀粉含量较高的原因。因此调控籽粒蔗糖代谢通路以及淀粉水解通路可以作为调控小麦加工品质的新思路。
