高效液相色谱法测定郁金香鳞茎中3种内源激素
2021-02-26赵杨静唐道城
赵杨静 唐 楠 唐道城 张 静
(青海大学高原花卉研究中心,青海省园林植物与观赏园艺重点实验室,西宁 810016)
植物内源激素是指在植物体内合成的、通常从合成部位运往作用部位对植物的生长发育起显著调节作用的微量生理活性物质[1~3],主要包括生长素类、赤霉素类、细胞分裂素类、脱落酸和乙烯[4]。植物生长发育过程中,任何一种生理效应都是各类激素相互作用的结果,其中激素在植物的成花及其他生长发育中都起着关键性作用[5]。
郁金香(Tulipa gesneriana L.)为百合科(Lilia⁃ceae)郁金香属(Tulipa L.)多年生鳞茎类草本花卉,是世界上著名的观赏植物之一,广泛应用于切花、盆栽和庭院种植[6]。郁金香是典型的夏休眠球根花卉,需要经过一段时间的低温处理才能打破休眠,促进花茎发育和伸长[7~8]。王晓东等对郁金香生长发育和内源激素含量进行了研究,实验结果皆表明内源激素对郁金香的花芽分化,生长发育起着重要的调节作用[9]。这种对环境的反应及花茎发育与伸长基因的表达均受自身内源激素的调控,所以鳞茎体内内源激素的分布规律和含量的准确测定,对研究植物生理变化及栽培应用都具有重要意义[10]。
目前,测定植物内源激素的方法主要有高效液相色谱法(HPLC)[11]、液相色谱串联质谱法(HPLC-MS)[12]、气相色谱串联质谱(GC-MS)及免疫分析法(ELISA)等[13]。田如男、魏钰等用ELISA法对郁金香球内的内源激素进行了研究,试验得出该方法具有操作简单、快速、敏感性高的特点,但重现性差、易交叉感染、不能同时分析多种激素,在精确定量测定方面存在不足[2,14~15]。GC-MS法广泛应用于有机物的现场分析检测,定量分析精度高,定性能力高,但维护成本高,结构复杂,在实验室使用时要经过专门培训的技术职员才能操纵质谱仪[16]。HPLC-MS 法可以直接分析激素,具有分析速度快,定性分析结果可靠,但仪器的高成本阻碍了其更广泛的应用[14]。HPLC 法是除了乙烯以外,其他内源激素均可测定的方法[17]。侯凯、李华、范光宇等均用HPLC 法测定川白芷、枇杷果实和谷子叶尖组织中的内源激素的含量[18~20],结果表明,该方法广泛应用于植物内源激素的测定,具有简单快捷,费用低、重现性较好等优点。
HPLC 法也应用于百合科植物内源激素的检测[21],但在测定郁金香鳞茎内源激素含量和程序优化这一方面研究报导较少。本研究以郁金香鳞茎为材料,通过对高效液相色谱条件和提纯方法的选择,筛选出能高效测定郁金香鳞茎内GA3、IAA 和ABA 3 种内源激素的方法。本方法灵敏度高、重现性好,能够满足郁金香鳞茎主要内源激素的定量分析,为郁金香鳞茎激素变化规律的研究供技术支持。
1 材料与方法
1.1 仪器、试剂与材料
高效液相色谱仪(Agilent 1260,配UV 检测器)、C18 固相色谱柱(5 μm,4.6 mm×150 mm Agi⁃lent ZORBAX SB-C18)、氮吹仪(上海海科DCY-12G)、冷冻离心机(SIGMA 3-30K)、冰箱(AUCMA BCD-239EG)、摇床培养箱(新苗全温培养摇床QYC-200)、超纯水制备系统(优普超纯水制造系统)、超低温冰箱(Thermo scientific)、Eppendorf 移液器、万分之一天平(上海良平FA1004)。
赤霉素(GA3,纯度≥98.6%)、吲哚-3-乙酸(IAA,纯度≥99.3%)、脱落酸(ABA,纯度≥98.5%)标准品购自Sigma Aldrich,异丙醇(分析纯)、盐酸、二氯甲烷(分析纯)、磷酸(分析纯)、甲醇(色谱纯)。
试验以青海大学高原花卉研究中心自繁的郁金香品种“Golden Apeldoorn”为材料,选取周径8~10cm,基盘无损伤且无病虫害的鳞茎。
1.2 实验方法
1.2.1色谱条件
色谱柱:Agilent ZORBAX SB-C18;流动相:A(甲醇)∶B(磷酸缓冲液pH=3.5)=45∶55;流速1 mL·min-1;检测 波长:0~3.2 min 265 nm,3.0~4.5 min 212 nm,4.5~6.5 min 218 nm,6.5~13.0 min 265 nm;自动进样,进样量10 μL;柱温20℃。
1.2.2标样制备
分别标准称取1 000 μg GA3、IAA、ABA 标准品溶于80%甲醇中配制成1 000 μg·mL-1的溶液,最后稀释成7 个浓度梯度,过0.45 μm 有机针孔滤膜,置于-80℃超低温冰箱备用(见表1)。
1.2.3样品前处理
取郁金香鳞茎,置于冰块上迅速切碎混合,放入研钵,液氮中研磨成粉末。
异丙醇提取方法:称后取0.5 g鳞茎粉末,加入5 mL 提取液A(超纯水∶异丙醇∶盐酸=1∶2∶0.002,V/V/V),4℃摇床100 r·min-1处理30 min,加入8 mL提取液B(二氯甲烷),4℃摇床100 r·min-1处理30 min;取出后4℃5 000 r·min-1离心15 min,此时溶液分层,粉末状组织呈絮状,位于两层溶液之间;用巴氏滴管吸取下层溶液,氮气吹干,加入1.0 mL 流动相溶解;过0.45 μm 有机针孔滤膜,装入样品瓶中置于4℃冰箱待测。

表1 混合标准品的7个浓度梯度Table 1 Seven concentration gradients of mixed standards
80%甲醇提取方法:称取鳞茎粉末1.0 g,加入10 mL 预冷80%甲醇,4℃冰箱过夜;4℃5 000 r·min-1离心15 min,取上清液,残渣用5 mL 80%甲醇清洗,离心合并上清液;氮气蒸发至原体积的1/3,调pH 至8.0,加入0.2 g PVPP,充分摇匀,冰箱放置30 min,离心,取上清液调pH至3.0,乙酸乙酯等体积混合萃取3次,合并3次萃取液,氮气吹干,1 mL 流动相溶解,过0.45 μm 有机针孔滤膜,装入样品瓶中置于4℃冰箱待测。
2 结果与分析
2.1 流动相初选
设置7 个流动相选择,其他条件均一致(流速为1.0 mL·min-1,进样10 μL,检测波长254 nm,柱温20℃),进行流动相的选择(见表2)。七个流动相对混合标准品GA310 000 ng·mL-1,IAA 2 000 ng·mL-1,ABA 10 000 ng·mL-1进行检测,积分标准为斜率灵敏度0.05,峰宽0.02,最小峰面积0.05,分离度大于1.5为完全分开(见表3)。
测定过程中按各激素的保留时间为顺序,分别 为GA3、IAA 和ABA。流 动 相1 测 定 时 间 为30.041 min,测定时间过长;流动相3、4、7的测定时间分别为4.534、4.326和4.733min,测定时间过短,若样品中杂质未去除干净,可能会与GA3峰重叠,从而导致分离效果差;流动相2、5、6检测时间分别为10.349、17.377、8.824 min,检测时间较适合,分离度和对称因子较高,其中流动相5峰宽较宽,专一性会降低,因此初步选取的流动相为2、6(见图1)。
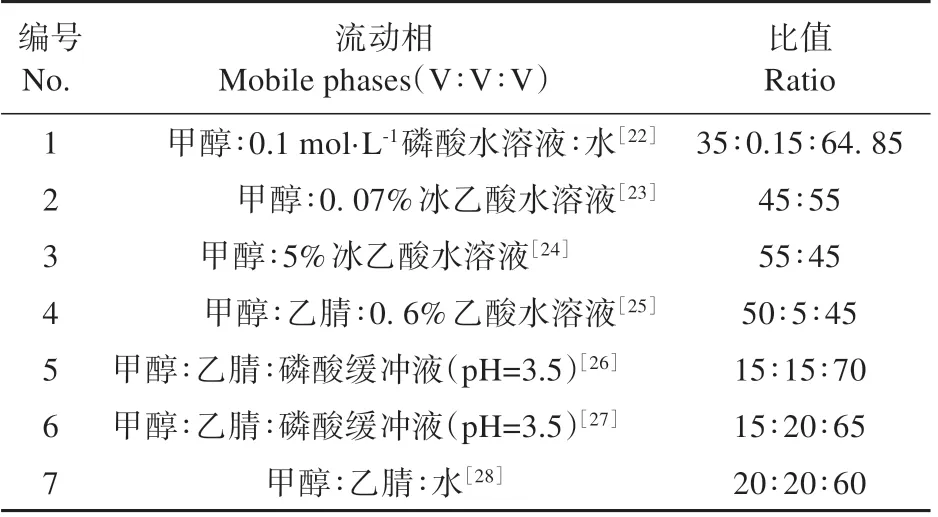
表2 7个流动相的配比Table 2 Ratio of seven mobile phases
2.2 波长选择及流动相改进
由流动相初选可知,254 nm波长下GA3和IAA峰面积小,不利于更低量的测定,所以需对测定波长进行选择。GA3的最大紫外吸收波长为210 nm[29],ABA 最大吸收波长为265 nm[30],通过对标准品IAA 的不同波长测定(见表4),可得IAA 的最大吸收波长为218 nm。
由于甲醇的截止波长为210 nm,在210 和218 nm 噪声较大色谱基线不稳定,而在265 nm 处基线噪音更小,所以除测定激素时间外,其他时间选用265 nm,即切换波长时间设定为0~3 min 265 nm,3.0~3.5 min 212 nm,3.5~5.2 min 218 nm,5.2~12.0 min 265 nm。切换波长测定中,因为冰乙酸的截止吸收波长为230 nm,流动相2 在低波长210 nm 和218 nm 处基线不稳定,所以用磷酸(截止波长210 nm)代替冰乙酸,调pH=3.5,分别用改良流动相2 和流动相6 切换波长对标准品进行检测,二者均可得到良好的色谱信号,由于乙腈价格较甲醇高,本试验选择改良流动相2,以甲醇和磷酸缓冲液(pH=3.5)以45:55(V/V)作为流动相。

表3 7个流动相色谱参数Table 3 The chromatographic parameters of seven mobile phases
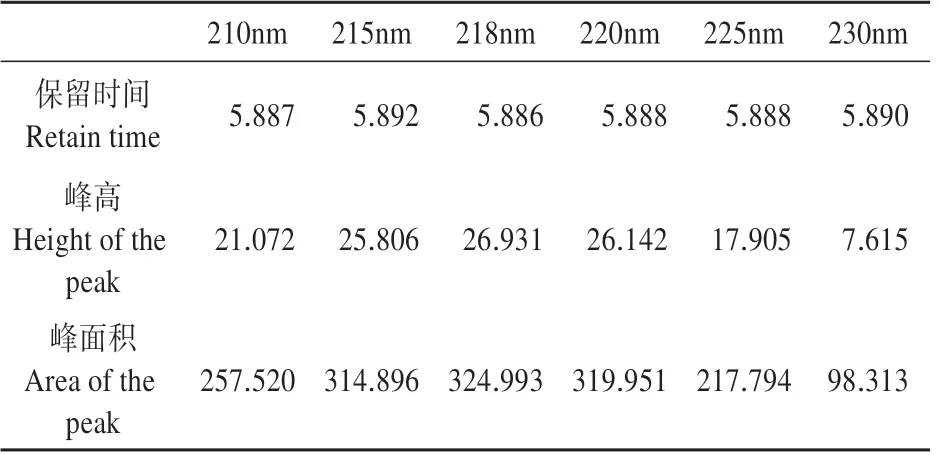
表4 HPLC法不同波长IAA色谱峰参数Table 4 Chromatographic parameters of IAA in differ‐ent wavelengths
2.3 柱温和流速的选择
柱温设定为20℃、25℃、30℃、35℃,结果表明,柱温对色谱分离影响不大,主要是影响保留时间和压力,柱温越高,保留时间越短,柱内压力越小,但考虑GA3分离时间不能提前,且高温不利于激素保存,所以本试验选择20℃为检测柱温。
流速设定0.8、1.0、1.2 mL·min-1,结果表明流速对各激素峰形影响不大,流速越大,分离时间越短,柱压越大,综合时间和柱压考虑,流速选用1.0 mL·min-1。
2.4 内源激素测定方法分析
2.4.1定性分析
利用以上优化条件,对标准品混合样GA310 000 ng·mL-1,IAA 2 000 ng·mL-1,ABA 10 000 ng·mL-1进行测定,3 种植物激素保留时间依次为3.688 0±0.002 min、5.794±0.002 min、11.324±0.004 min(见图2)。
2.4.2定量分析
将配制的7 个浓度梯度混合标准品进行HPLC 检测。以峰面积为纵坐标(y),含量为横坐标(x),得到各内源激素的线性回归方程及相关系数(见表5)。

表5 3种内源激素标准品的标准曲线Table 5 The standard curve of three endogenous hor‐mone standards
2.5 样品处理方法的选择
本试验用异丙醇[4]和80%甲醇[2,15,31]作为提取试剂。两种方法均能提取检测出3 种激素(见图3),经定量检测80%甲醇提取方法测定激素含量分 别 为:GA3(103.549±3.690)ng·g-1FW,IAA(11.378±2.050)ng·g-1FW,ABA(136.451±4.880)ng·g-1FW;异丙醇提取剂提取方法测定激素含量分 别 为:GA3(1 140.232±23.686)ng·g-1FW,IAA(20.145±2.979)ng·g-1FW,ABA(117.733±19.395)ng·g-1FW。定量对比可得,80%甲醇作为提取剂进行样品提取GA3损失较多,综上所述,选择异丙醇提取方法作为郁金香鳞茎激素提取方法。
2.6 回收率测定
标准品母液精确取10 000 ng GA3,2 000 ng IAA,10 000 ng ABA 加入5 mL 异丙醇提取液试管中,按照样品处理方法处理标准品,HPLC 进行6次平行检测,计算回收率。3种激素回收率分别为:GA389.190%,IAA 84.821%,ABA 95.679%,相对标准偏差依次为:6.432%,4.373%,2.831%(见表6)。

表6 3种内源激素的回收率及相对标准偏差Table 6 Recoveries and RSDs of three endogenous hor‐mones
3 讨论
植物内源激素的提取是定量检测的第一步也是关键步骤,样品的处理方法主要有液氮研磨[20]、冰浴研磨[32]、常温研磨[33]3 种,本试验结合实验室条件,为了最大程度保持激素活性,最终采用液氮研磨样品的方法。目前,植物激素的提取方法多采用溶剂提取法,常用溶剂有甲醇、丙酮、丙醇水溶液及其他中性酸性缓冲溶液[34~35],因甲醇分子量小,植物激素提取中多采用甲醇作为提取剂[22~24],但甲醇渗透性极强,选择性低,易将植物细胞内的多酚类物质和植物内源激素一并提取出来,故在测定植物内源激素前需要通过一定的纯化步骤,尽可能地将这些杂质除去,这就导致后续工序复杂,工作量加大,提取率降低。郁金香鳞茎的多酚类物质含量较高[36],为简化后续试验步骤,保证较高的提取效果,因此本研究选择异丙醇溶液作为郁金香鳞茎内源激素提取剂。
国内有大量HPLC 法测定植物激素的方法报道,但因仪器、分离柱及温度、气压等因素的差异,各实验室分离条件参数相差较大。本方法参考国内常见植物内源激素的样品前处理方法和色谱测定参数,从样品研磨方法、样品激素提取剂的选择,流动相、流速、柱温、吸收波长等方面进行了优化,最终筛选出一套适合实验室高效液相色谱仪同时测定郁金香鳞茎中3种植物内源激素的方法。该方法以液氮研磨样品,异丙醇为激素提取剂,Agilent1260 高效液相色谱仪为检测仪器,色谱柱Agilent ZORBAX SB-C18;流动相A(甲醇)∶B(磷酸缓冲液pH=3.5)=45∶55;流速1.0 mL·min-1;检测波长0~3.2 min 265 nm,3.0~4.5 min 212 nm,4.5~6.5 min 218 nm,6.5~13.0 min 265 nm;自动进样,进样量10.0 μL;柱温20℃,且均在各激素最大吸收波长进行定量测定,基线平稳,准确度高,可得到满意的峰形,色谱图规范整齐,试验前处理简单,可操作性强,回收率和精密度较高,可满足测定郁金香鳞茎GA3、IAA、ABA含量的要求。
