基于密度泛函理论对咔咯-吩噻嗪二元体激发态电荷转移的研究
2020-07-01高龙江刘海洋
高龙江, 刘海洋
(华南理工大学化学系, 广州 510641)
太阳能是一种重要的可再生能源,光合作用是植物和其他生物体利用光合色素将太阳能转化为化学能的过程,涉及光能吸收和电荷分离[1-2]. 为了解释光合作用机理,人们已经在人工光合作用模型系统的构建上投入了大量精力,其中,较常见的做法是构建电荷供体-受体体系,人工模拟光合作用产生的过程,对供受体种类、供体取代位置和间隔基进行研究[3-5].
咔咯是具有18π的类卟啉大环化合物[6],咔咯环上中位和β位可被修饰,使其衍生物多样化. 由于与叶绿素的相似性,咔咯作为一种供体-受体系统中常见的受体材料[7-9]. 吩噻嗪(phenothiazine,PTZ)是一种三环杂芳族发色团,由于同时存在给电子的N原子和S原子,在研究中PTZ常被作为电子供体[10-11]. 非平面且具有蝶形构象是PTZ的重要结构特征,这些特征会阻碍分子聚集和分子间准分子的形成.
本文利用5,15-二(五氟苯基)咔咯(F10C)与受体PTZ构建7种二元体作为供体-受体模型,使用密度泛函理论方法分别从几何结构、前线分子轨道、电子-空穴分析、吸收光谱等方面对咔咯-吩噻嗪二元体的供体-受体电荷转移进行了计算研究,探讨了供体的位置和间隔基的性质对供体-受体激发态电荷转移的影响.
1 计算方法
研究了F10C、PTZ和7种二元体等9种分子(图1),所有计算均用Gaussian 09程序[12]完成. 采用极化连续介质模型(Polarizable Continuum Model,PCM)计算CH2Cl2的溶剂效应. 采用B3LYP/6-31G(d,p)基组,对分子进行基态几何构型优化. 采用TD-DFT理论[13-14]对PTZ进行激发态计算,方法和基组为B3LYP/6-31G(d,p). 其他分子采用CAM -B3LYP/6-31G (d,p)基组进行激发态计算. 采用Multiwfn程序[15]和VMD程序[16]对结果进行分析.

图1 PTZ、F10C、F10C-PTZ二元体的分子结构
2 结果与讨论
2.1 分子构型
对图1中的分子进行结构优化后得到如表1所列的数据. 二元体1中供体和受体直接相连,二元体2和3中供体和受体分别通过间隔基(C—C键和CC键)连接. 二元体4~7是供体直接连接在受体的不同β位(β1~4)上所形成的二元体结构. 在二元体2中,由于间隔基未在供体和受体之间形成π-π共轭,间隔基与受体连接所形成C—C键的键长明显大于其它二元体,并且受体和供体之间形成的二面角(87.524°)明显小于其它二元体. 所有二元体的D1,S,N,2角度与单体PTZ几何优化结构中的该角度(136.574°)相比,差异极小,表明供体-受体体系对供体PTZ的蝶形结构无明显影响. 咔咯环具有近乎平面的芳香环结构,4个吡咯近似共面[17]. 二元体1~7中,DN,N,N,N角度均小于5°,所有二元体的咔咯环内平均C—N键长近似相等,说明供体取代基对受体的平面咔咯环没有造成明显的扭曲或伸缩.
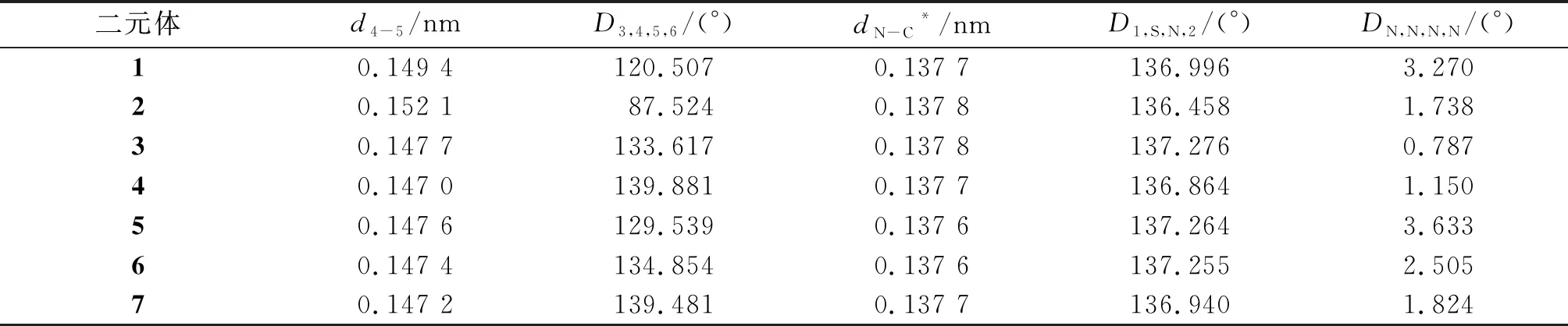
表1 F10C-PTZ二元体的优化几何结构参数Table 1 The optimized geometrical structure parameters of F10C-PTZ dyads
注:*表示平均值;d为键长;D为二面角.
2.2 前线分子轨道分析
图2显示了PTZ、F10C和二元体的最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO). 考虑到供体的LUMO轨道能级为-0.880 eV,F10C的LUMO能级比供体PTZ的LUMO低1.085 eV,因此推测在供体和受体之间存在电荷转移的激发态[18]. 7种二元体的HOMO和LUMO电子云主要集中在咔咯大环骨架上,并且电子云高度相似. 二元体1和3的HOMO有少量电子云分布在供体上. 供体和受体结合后,二元体1~7整体的HOMO和LUMO能级与F10C的相仿,说明这些供体-受体体系中,受体对体系的HOMO-LUMO轨道起主要作用.
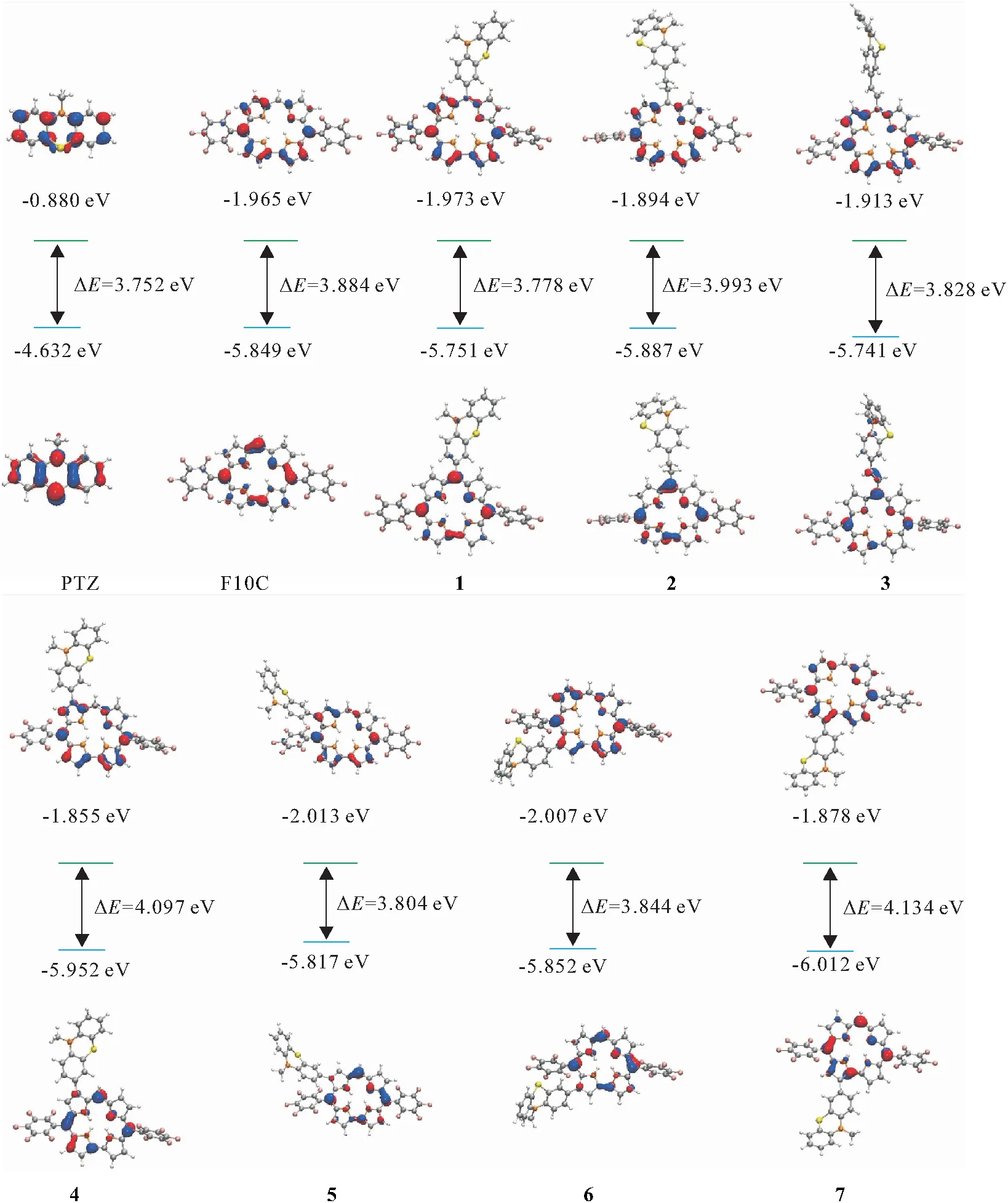
图2 PTZ、F10C、F10C-PTZ二元体的前线分子轨道及能级
2.3 二元体激发态分析
图3是二元体的模拟吸收光谱,受体F10C与供体PTZ在波长200~500 nm之间的吸收光谱有大面积重叠,这表明供体-受体体系在该区间有发生分子内电荷转移的可能性. 在重叠区间,二元体的激发态S3~S6起主要作用,所以本文计算二元体的激发态S1~S10,对二元体供体-受体电荷转移激发进行研究. 二元体1~7与受体F10C的模拟吸收光谱相似,都在波长400 nm和650 nm附近有2个明显的吸收峰,在波长400 nm附近的吸收峰最大. 对于波长650 nm附近的吸收峰,在二元体1~7中,S1是主要贡献的激发态,其中二元体4和7中的S2亦有明显贡献;对于波长400 nm附近的吸收峰,S3和S4是主要贡献的激发态,值得一提的是,在二元体5和6中,激发态S5有显著的贡献.

图3 F10C、PTZ和F10C-PTZ二元体的模拟吸收光谱
使用Multiwfn程序中的Interfragment Charge Transfer(IFCT)[19]方法,将二元体中F10C结构设为受体,将PTZ和间隔基设为供体,得到供体向受体F10C的净电荷转移量(表2). S4是二元体1和3开始发生供体-受体电荷转移激发(净电荷转移量大于0.1)时的激发态,S5是两者净电荷转移量最大时的激发态,分别为0.723 30 e和0.482 03 e. 二元体2从S5开始发生供体-受体电荷转移,同时S5也是该二元体中供体-受体净电荷转移量最大的激发态.
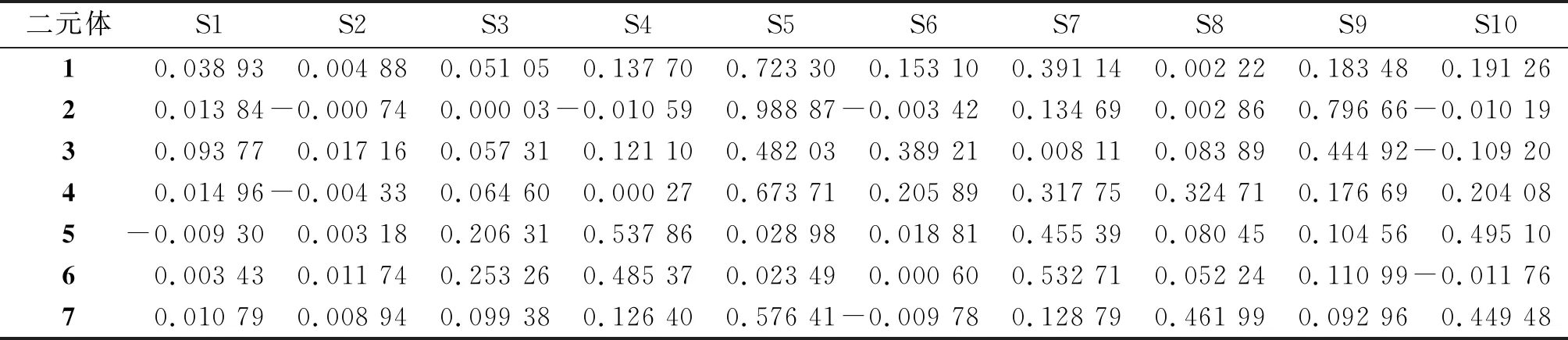
表2 F10C-PTZ二元体的S1-10的供体-受体净电荷转移结果Table 2 The donor-acceptor net charge transfer results for S1-10 of F10C-PTZ dyads e
注:负号表示受体到供体的电子转移.
二元体激发态的电子-空穴分布图(图4)可直观地分析激发态电子跃迁. 在二元体2的激发态S5中,空穴和电子独立地分布在供体和受体上,空穴-电子分布重叠程度低,供体-受体间净电荷转移量高达0.988 87 e(表2). 将二元体2和3对比,在二元体3中,间隔基CC键在受体和供体之间形成π-π共轭,电子和空穴均会分布在间隔基上(图4); 在二元体2中,间隔基C—C键几乎不参与电荷转移.
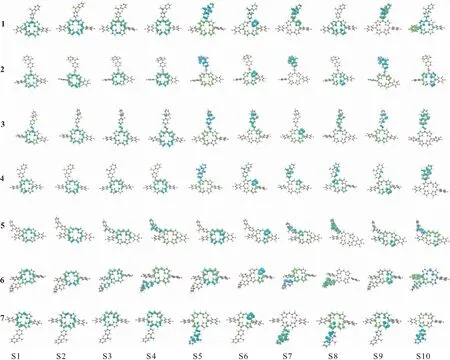
图4 二元体S1~S10的电子-空穴分布图
针对间隔基的考察表明,C—C键几乎不参与电荷转移激发,降低了供体和受体之间的相互作用,使电荷转移激发态减少,并且电荷转移激发过程发生在高激发态. 在二元体2中,除S5和S9以外,其他激发态的供体-受体净电荷转移量都明显小于二元体1和3. CC键导致体系共轭性的增加,间隔基会参与到电荷转移激发过程中,增强供体和受体之间的相互作用,电荷分布比较平均,不同激发态的净电荷转移量波动比较平缓.
二元体1、4、5、6、7分别是受体在供体不同位置的取代.β1位和β4位取代的二元体4和7在几何结构上与1的相近,三者具有相似的电子-空穴分布(图4). 表2显示二元体1、4、7三者的激发态净电荷转移量差别不大,其中,S5同时是二元体1、4、7发生供体-受体净电荷转移量最高的激发态,分别为0.723 30 e、0.673 71 e、0.576 41 e. 二元体5和6的供体取代位置β2位和β3位与咔咯环上的五氟苯基相邻,由于五氟苯基的拉电子效应,导致二元体5和6均从低激发态S3开始发生供体-受体电荷转移. 值得一提的是,二元体5和6的几何结构相似,两者的激发态S1~S9的电子-空穴分布高度相似,对应的激发态净电荷转移量差异很小.
3 结论
利用密度泛函理论对咔咯F10C和吩噻嗪PTZ形成的一系列二元体进行了激发态电荷转移研究. 结果发现:供体的取代位置和间隔基性质均对供体-受体电荷转移有显著影响. C—C键作为间隔基会降低供体-受体的相互作用,减少二元体供体-受体激发态电荷转移,电荷转移激发态出现在更高能级激发态;CC键可增加体系的共轭性,并参与到电荷转移激发中,电子和空穴均会在CC键上分布,增强供体-受体的相互作用. 对供体取代位置的考察表明,由于咔咯β1和β4位与中位相似,二元体1、4、7的电荷转移激发态相近.β2位和β3位取代的二元体5和6具有相似的激发态电荷转移,由于两者的PTZ与五氟苯基位置相邻,五氟苯基的拉电子效应会导致电荷转移激发出现在低能级激发态,有利于供体-受体电荷转移的发生.
