一例因基因突变致LIG4综合征病例的临床特点及分析
2020-06-09张潇潇顾浩翔吴蓓蓉
张潇潇,陆 敏,顾浩翔,吴蓓蓉
(上海市儿童医院 上海交通大学附属儿童医院 小儿呼吸内科, 上海 200040)
DNA连接酶Ⅳ(DNA ligase Ⅳ,LIG4)位于染色体13q33-34上,编码911个氨基酸的多肽[1]。 DNA连接酶Ⅳ突变导致LIG4综合征,这是一种极其罕见的常染色体隐性遗传免疫缺陷病,是由于DNA非同源末端连接(non-homologous DNA end joining,NHEJ)修复缺陷导致。其临床特征为小头畸形, 特殊面容,发育迟缓,血细胞数量减少和不同程度的免疫缺陷[2]。本文对一例LIG4综合征患儿的临床特点及基因检测结果进行总结,并结合相关文献进行分析。
1 材料与方法
1.1 对象
上海市儿童医院诊治LIG4综合征一例:患儿,男,1岁8个月,G1P1,足月顺产,出生史无异常。患儿父母非近亲结婚,否认家族性、遗传性病史。
本研究获得了上海市儿童医院伦理委员会的批准(批准文号:2016R025-E01),并得到了患儿家属的知情同意。
1.2 方法
二代高通量基因测序:采集患儿(及父母)静脉血2 mL,置乙二胺四乙酸(EDTA)抗凝管中,用QIAamp DNA Blood Mini Kit DNA提取试剂盒提取基因组DNA。用NANODrop 2000 (Thermo公司)及1.5%琼脂糖凝胶电泳对DNA质量和浓度进行检测。用超声仪将DNA随机打断成300~400 bp片段,纯化产物经末端修复后进行接头连接、PCR扩增,构建测序文库,使用HiseqX10(Illumia公司)平台进行测序。根据美国医学遗传学和基因组学学院/分子病理学协会(ACMG/AMP)的变异分类指南对统计出的变异结果进行致病性分类。对判断为致病、可能致病及临床意义未明的患者,采用相同变异位点的Sanger测序法对患者及父母进行检测。
2 结果
2.1 本例患儿临床特点
患儿,男,1岁8个月,因“咳嗽1月余,发热4 d”入住上海市儿童医院。既往有反复呼吸道感染、慢性腹泻病史,患儿及其家长从未去过南美洲。查体:T 36.7 ℃,P 120次/min,R 35次/min,体质量9.5 kg,身长84 cm,头围41 cm,。神志清,呼吸平稳,反应可,小头畸形,三角形脸,眼睛轻微下斜,鼻翼较宽,全身皮肤黏膜未见皮疹及出血点,浅表淋巴结未及明显肿大,吸凹(-),无鼻翼扇动。睑结膜无充血,颈软,咽部充血,口腔有大量白絮状物,扁桃体I度肿大,胸廓对称,无畸形,两侧呼吸运动对称,肋间隙未见明显增宽。双肺叩诊呈清音,双肺呼吸音粗,可闻及痰鸣音。心音有力,律齐,未及杂音,腹软,不胀,无压痛,无反跳痛,四肢活动可,肛门生殖器无异常,神经系统查体未见异常。
实验室检查:血常规示WBC 3.53×109/L,RBC 4.08×1012/L,Hb 112 g/L,PLT 353×109/L,N 0.56,L 0.20,CRP 30 mg/L,网织红细胞1.36%。肝肾功能电解质正常。病原学:呼吸道合胞病毒(痰液),耐药支原体感染(肺泡灌洗液DNA)、流感嗜血杆菌+白色念珠菌(肺泡灌洗液),EB病毒(DNA),白色念珠菌(粪便)。细胞免疫、体液免疫功能缺陷(表1)。
其他检查:支气管黏膜活检:电镜下大量脱落黏膜柱状上皮细胞,表面微绒毛丰富,部分上皮细胞溶酶体增多。视野中所有细胞均未见纤毛结构。超微结构观察提示:黏膜上皮细胞变性、脱落,纤毛缺失可疑 (图1)。
2.2 基因检测结果
基因检测发现本例患儿的LIG4基因第833位碱基存在纯合性改变:c.833G>T(p.R278L),分别遗传自患儿父母(图2)。
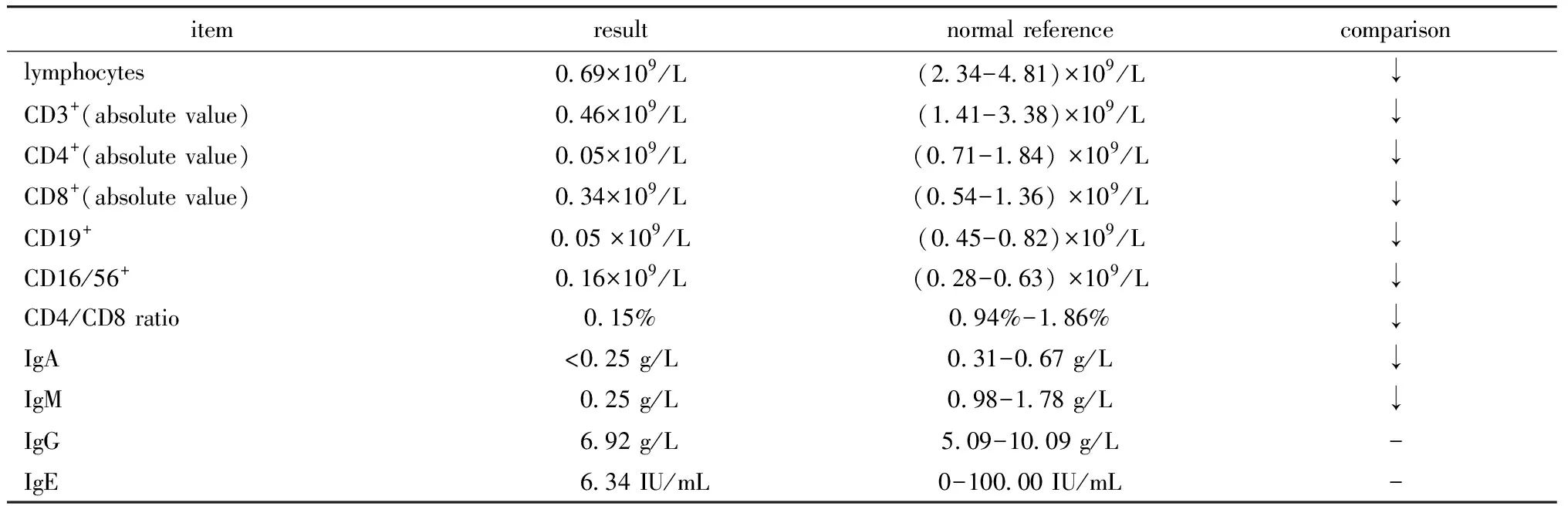
表1 本例患儿免疫功能检查结果Table 1 The child’s immune function test results
A.a large number of mucosal columnar epithelial cells were shed, and the surface was rich in microvilli, no ciliary structure was seen in all cells in the visual field; B.mucosal epithelial cells degenerated, shed, and cilia were deleted
图1 患儿支气管黏膜活检电镜结果
Fig 1 Electron micrograph of bronchial mucosal biopsy of the child
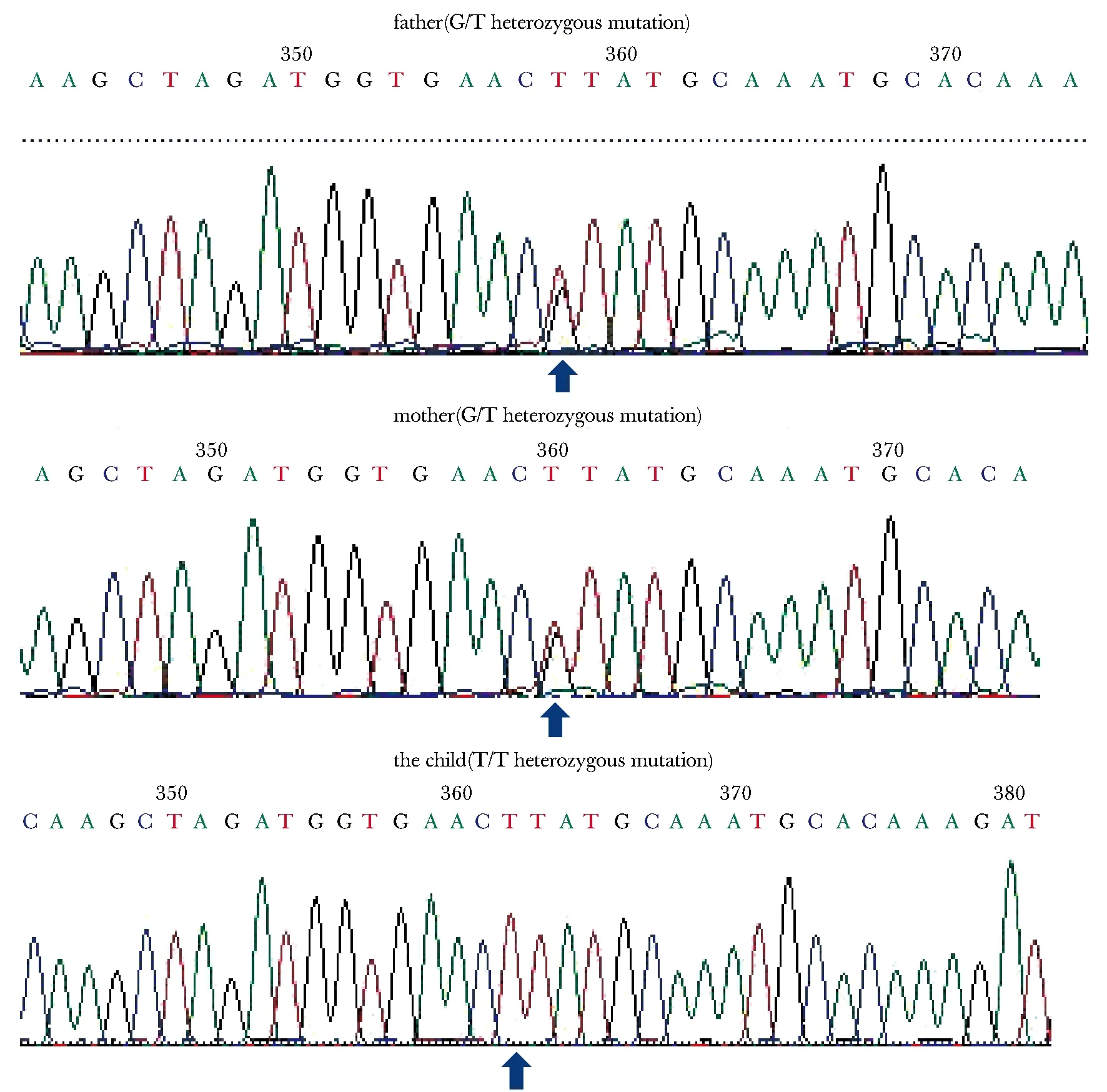
the arrow showed the mutation site图2 患儿及其父母的LIG4基因测序结果Fig 2 Sequencing results of LIG4 gene of the child and his parents
3 讨论
DNA双链断裂(DNA double strand break,DSB)是最常见的DNA损伤形式,这些病变可由正常的内源性过程诱导,包括DNA复制、减数分裂和B(T)细胞发育期间的V(D)J重组[3],以及外源性的电离辐射(IR)、细胞毒性药物。DNA修复缺陷可导致细胞死亡、基因突变和癌症。DNA修复可通过非同源末端连接(NHEJ)或同源重组(homologous recombina-tion,HR)修复。NHEJ是主要的DSB修复途径,其中两个末端在限制性内切酶处理后重新连接。至少6种蛋白质组分在NHEJ中起作用,包括:Ku70、Ku80、DNA-PKcs(蛋白激酶DNA激活催化多肽,PRKDC)、DNA连接酶Ⅳ、XRCC4(X线修复交叉互补蛋白4)和Artemis[4],其中DNA连接酶Ⅳ对于进行最终的“末端连接”必不可少。DSB修复缺陷可导致癌症易感性、免疫功能障碍、放射敏感性、发育迟缓和神经变性。NHEJ介导的DSB修复缺陷占人类遗传性联合免疫缺陷病的15%。通过PubMed、Springer Link和Elsevier数据库、万方数据库、CNKI数据库检索出LIG4综合征病例42例(包括本研究一例)。LIG4综合征临床特征主要有:小头畸形42例(100%),不同程度免疫缺陷42例(100%),特殊面容21例(50%),血细胞下降17例(40%),发育迟缓16例(38%),放射敏感性11例(26%),肿瘤9例(21%)。其中4例造血干细胞移植成功,其余均死于感染、移植后排异反应等。目前共发现25种基因突变类型(表2),其中c.833G>T(p.R278L)是中国LIG4综合征患者的突变热点。预测R278L可能通过改变氢键来影响DNA连接酶Ⅳ的蛋白质结构[5]。本例患者基因检测结果即为c.833G >T纯合子突变。
免疫缺陷是LIG4综合征患者主要的临床表现之一。B细胞和T细胞分化的早期阶段产生抗原特异性B细胞和T细胞受体,V(D)J重组缺陷导致免疫缺陷。V(D)J重组过程中产生发夹密封的编码末端和钝化的磷酸化信号末端。这些DNA末端被NHEJ途径的蛋白识别和分析,分别形成编码连接和重组信号。KU70/KU80异二聚体直接结合DNA末端,允许激活DNA-PK催化亚基(DNA-PKcs)和磷酸化Artemis, 在NHEJ的最后阶段, DNA连接酶Ⅳ(LIG4)/XRCC4复合物介导DNA末端的连接[12]。受损的V(D)J重组,T和B淋巴细胞生成中的缺陷导致B和T辅助性淋巴细胞减少症。不同程度的免疫缺陷可能是由于有些类型的突变产生少量正常的活性蛋白与低于检测下限的LIG4蛋白[13]。低水平的LIG4蛋白足以使细胞存活,但V(D)J重组显然需要更高的LIG4蛋白水平。本例患儿存在体液免疫及细胞免疫缺陷,导致反复的呼吸道感染,长期的抗生素治疗破坏了内环境的紊乱,继发真菌感染,胃肠功能紊乱。

表2 文献报道的LIG4基因突变Table 2 Mutations of LIG4 gene from literatures
Murray 等[8]报告了11例小头畸形患者的LIG4基因突变,所有患者父母都是正常表型的杂合子携带者,患者表现出严重的产前和产后发育迟缓。这项研究扩展了与LIG4突变相关的表型谱即小头畸形、发育迟缓是LIG4综合征的常见表现。Rucci等[14]研究发现,在LIG4缺陷小鼠中,大量神经元细胞凋亡,是导致小头畸形、发育迟缓的主要原因。很多LIG4综合征患者在儿童早期有相似的面容,常见特征有毛发稀疏,赘肉褶皱,宽鼻窦,鼻尖宽,下巴突出,也称作“鸟样面容”[8]。LIG4综合征患者存在血细胞下降,其中血小板受影响最严重,其次是白细胞,血红蛋白仅轻度降低。骨髓穿刺没有发现形态异常的幼稚骨髓细胞,随着年龄的增长需要输注血制品的次数增加,逐渐进展为骨髓衰竭[15],骨髓衰竭的原因是由于DSB在多能造血干细胞发育过程中逐渐积累导致细胞凋亡[16]。一例LIG4基因突变的急性淋巴细胞白血病患者,因预防性头颅放射治疗致头皮脱落和双侧乳突放射性溃疡脱落,并死于放射性脑病[17]。从LIG4综合征患者体内分离出的人成纤维细胞放射敏感性增加[18]。此外,部分LIG4综合征患者出现恶性肿瘤,Bacon等[19]报道了 2例患者有EB病毒相关淋巴瘤,还有恶性鳞状细胞癌的报道[18]。
值得注意的一点是,LIG4基因缺陷的临床和免疫表现是可变的。已经报道了一些患者中有自发性体细胞修复的情况,这种现象称为“天然基因疗法”[20],可使患者存活时间延长。推测有可能是重组介导的基因内交叉引起体细胞的修复。自发性逆转患者的疾病进程有所改善,如生存期延长和症状减轻,但这些改善不足以预防致命性并发症,并且无法完全恢复细胞功能,远期临床进展和机制尚不清楚。因此需要更多的案例和研究来揭示这种罕见疾病的罕见情况。这种现象或许将来可以为基因治疗提供思路。
