Folic acid attenuates high-fat diet-induced steatohepatitis via deacetylase SlRT1-dependent restoration of PPARα
2020-06-08FengZhiXinZeHuaZhaoRuiNanZhangQinPanZiZhenGongChaoSunJianGaoFan
Feng-Zhi Xin, Ze-Hua Zhao, Rui-Nan Zhang, Qin Pan, Zi-Zhen Gong, Chao Sun, Jian-Gao Fan
Abstract
Key words: Nonalcoholic fatty liver disease; Folic acid; Gut microbiota; PPARα; SIRT1
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) has become one of the main causes of chronic liver disease worldwide[1]. The prevalence of NAFLD in China has increased from 18% to 29% in the past ten years[2,3]. Similar trends have been observed in other parts of world. Non-alcoholic steatohepatitis (NASH), which is a subtype of NAFLD,increases the risk of cirrhosis, hepatocellular carcinoma, and liver-related death[4].However, there are still no drugs approved for treatment of NASH[5]. Therefore,NAFLD has become a serious global health burden and it is critical to find new drug targets for treatment of NASH.
Folic acid is an important substrate for the synthesis of methyl donors as an essential water-soluble vitamin metabolized by the intestinal flora and the human body[6]. Dietary folic acid could be absorbed and metabolized through the small intestine and liver. Finally, 5-methyltetrahydrofolic acid (5-MTHF) is the active form in blood circulation[7]. Folic acid deficiency could induce hyperhomocysteinemia and NAFLD. Dietary folic acid is essential for whole body folate homeostasis[8]. Additional folic acid supplementation could attenuate liver injury under high-fat diet (HFD)-fed or binge drinking conditions[9,10]. Dietary folic acid has been shown to ameliorate liver lipid accumulation[11-13]. All present data indicates that folic acid may become a potential drug target for treatment of NASH. However, further molecular mechanisms of folic acid on hepatic lipid and one-carbon unit metabolism are still unclear. The effect of folic acid on gut microbiota in NASH is also unknown. Taken together, it is necessary to further access the effect of folic acid on NASH and its possible mechanism.
To address the problems mentioned above, we conducted this research in HFDinduced NASH rats and palmitic acid (PA)-treated Huh7 cell line. Liver histology,hepatic one-carbon metabolism, and gut microbiota were evaluatedin vivoto investigate the effect of folic acid in NASH. Genes related to lipid metabolism were evaluated bothin vivoandvitroto illustrate the role of folic acid in hepatic lipid metabolism in NASH.
MATERIALS AND METHODS
Animal experiments
The animal experiments were performed in a way that discomfort for animals was minimized. A total of 24 six-week-old specific-pathogen-free (SPF) male Sprague-Dawley rats (Sippurbec Laboratory Animal Co., Ltd., Shanghai, China) were fed in a controlled environment (24 ± 1 °C, 50% ± 5% humidity, 12-h light-dark cycle, free access to water and standard chow diet). After 1 wk of adaptive feeding, the rats were fed a chow diet or HFD (88% standard diet, 10% lard, and 2% cholesterol) for 8 wk.Then, rats fed an HFD were randomly divided into two groups and fed folic acid (15 mg/kg·d) or saline by gavage once daily for 8 wk. All rats were fasted overnight and then euthanized with pentobarbital sodium at the end of 16 wk.
All animal experiments followed the National Research Council's Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of SHRM (SHRM-IACUC-015).
Gut microbiota analysis
Fecal samples from rats were collected immediately upon defecation and then stored at -80 °C after being snap frozen in liquid nitrogen. Total fecal DNA was extracted using a TIANamp DNA Kit (Tiangen, Beijing, China) according to the manufacturer's protocol. The quality and quantity of DNA were verified with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, United States) and agarose gel. Extracted DNA was diluted to a concentration of 1 ng/µl and stored at -20 °C until further processing. The V4-V5 variable regions of 16S rRNA genes were amplified with universal primers 515F and 907R for bacterial diversity analysis.Amplicons were purified with the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, United States) and quantified using QuantiFluor™-ST(Promega, Wisconsin, United States) according to the manufacturer's instructions.Equal amounts of purified amplicon were pooled for subsequent sequencing. Raw sequencing data were given in FASTQ format. Paired-end reads were preprocessed using Trimmomatic software. Clean reads were subjected to primer sequence removal and clustering to generate operational taxonomic units using Vsearch software with a 97% similarity cutoff. All representative reads were annotated and blasted against the Silva database using the Ribosomal Database Project classifier (confidence threshold was 70%).
Histological analysis
The body weight and liver mass were recorded after the rats were euthanized.Approximately 1.0 cm × 1.0 cm × 1.5 cm liver tissues were fixed in 4%paraformaldehyde for hematoxylin-eosin (HE), Masson, and Sirius red staining.Approximately 1.0 cm × 1.0 cm × 1.0 cm liver tissues were snap frozen in liquid nitrogen and then frozen at -80 °C for oil red O staining. The other liver tissues were stored at -80 °C for further analyses. Steatosis (S), activity (A), and fibrosis (F) (SAF)score was used for analyzing hepatic histological alterations[14]. Approximately 0.5 cmlong sections of the terminal ileum were gently rinsed with phosphate-buffered saline and then fixed in 4% paraformaldehyde for HE and immunohistochemical staining.
Serum and tissue assays
Serum was obtained by centrifugation of whole blood at 3000 r/min at 4 °C. Serum folic acid, alanine aminotransferase (ALT), aspartate aminotransferase (AST), fasting blood glucose (FBG), triglycerides (TG), total cholesterol (TC), high-density lipoprotein (HDL), low-density lipoprotein (LDL), total bile acid (TBA), and homocysteine (Hcy) were measured with an automated analyzer (Sysmex CHEMIX-180, Japan). The liver TG and cholesterol levels were measured with assay kits(Applygen Technologies Inc., Beijing, China). Samples and the standard curve were measured according to the manufacturer's instructions.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from liver tissue using TRIzol (D9108B, Takara, Dalian,China). The concentration and purity of RNA samples were assessed on a NanoDrop 2000 spectrophotometer (Nanodrop Technologies). Total RNA (1000 ng) was converted to cDNA with RT master mix (RR036A, Takara, Dalian, China). Real-time quantitative polymerase chain reaction (qRT-PCR) was performed with the Applied Biosystems Vii7 with SYBR®Green Master Mix (Low Rox Plus) (11202ES08, YEASEN,Shanghai, China). The primer sequences are shown in Table 1. The specificity of the primers was determined by dissociation curves using Vii7 system SDS software.RPS18 (B661201-0001, Sangon Biotech) was used as the internal control. The 2-ΔΔCTmethod was used to analyze relative gene expression.
Western blot analysis
Protein levels of methionine adenosyltransferase 1A (MAT1A), silence information regulation factor 1 (SIRT1), peroxisome proliferator-activated receptor alpha (PPARα),carnitine palmitoyltransferase 1A (CPT1α), and fatty acid binding protein 1 (FABP1)in rat liver and SIRT1 and PPARα in the Huh7 cell line were determined by Western blot analysis. Briefly, liver proteins (45 µg) and cell proteins (15 µg) were separated by 8%, 10%, or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and then proteins were transferred from the gel to a polyvinylidene fluoride (PVDF) membrane under constant current, cold conditions. The membranes were blocked with Quickblock buffer (P0252, Beyotime, Shanghai, China) for 25 min at room temperature and were then incubated with primary antibody overnight at 4 °C. Primary antibodies include anti-MAT1A polyclonal antibody (AB217005, Abscitech, United States), anti-SIRT1 monoclonal antibody (189494, Abcam, Cambridge, United Kingdom), anti-PPARα polyclonal antibody (A6697, Abclonal, Wuhan, China), anti-CPT1α polyclonal antibody (128568, Abcam), anti-FABP1 polyclonal antibody (A5311, Abclonal), and anti-GAPDH monoclonal antibody (#5147, CST, United States). Horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse IgG (Beyotime) were used as secondary antibodies, and the membranes were incubated at room temperature for 1 h. Protein bands were detected using a Western chemiluminescent HRP substrate(Millipore Corporation, Billerica, MA, United States).
Cell culture and transfection
The Huh7 cell line was obtained from American Type Culture Collection (ATCC;Manassas, VA, United States) and was cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM; HyClone) supplemented with 10% fetal bovine serum(Gibco, CA, United States). PA powder (Sigma, St. Louis, United States) was dissolved in 1% fatty acid-free BSA (Sigma, St. Louis, United States) Milli-Q water at 70 °C and filtrated with a 0.22 μm filter. The concentration of the stock solution was 5 mmol/L.The concentration of working PA solution was 0.3 mmol/L. The intervention included 0.3 mmol/L PA and 1 or 10 µg/mL 5-MTHF (Sigma-Aldrich, United States).Briefly, after 12 h of serum-free treatment, cells with 5-MTHF or the same amount of phosphate-buffered saline were incubated as pretreatment for 12 h, and then cells were incubated in PA with or without 5-MTHF for another 12 h. The proteins were isolated according to the manufacturer's instructions.
The Huh7 cell line was transfected with 50 nmol/L SIRT1 siRNA (Genomeditech,Shanghai, China) or its negative control (NC; Genomeditech) with Lipofectamine 3000(Invitrogen, Carlsbad, United States) in Opti-MEM medium (Gibco, CA, United States). After 18 h, the medium was replaced by high-glucose DMEM without fetal bovine serum. Pretreatment and intervention were performed 24 h after transfection.
Statistical analysis
All the data are expressed as the mean ± SE. The results were analyzed using twotailed Student'st-test between two groups and one-way ANOVA followed by Dunnett's test among multiple groups. Nonparametric tests were used for discontinuous data.P< 0.05 was considered statistically significant. All the statistical methods mentioned above were reviewed by Guang-Yu Chen from the Clinical Epidemiology Center, Shanghai Jiao Tong University.
RESULTS
Folic acid ameliorates histological alterations in HFD-induced NASH independent of affecting body weight
After a 16-wk experimental period, all rats in the HFD group developed typical NASH characteristics. Body weight (Figure 1A) and liver index (Figure 1B) were significantly elevated in the HFD group compared with the control group.Administration of folic acid had no effect on body weight or epididymis fat of rats(Figure 1A and C). But it ameliorated HFD-induced NASH hepatic lesions in rats. As shown in Figure 1B, liver index showed a certain reduction in the folic acid group compared with the HFD group. Additionally, folic acid improved the liver imaging results to a certain extent and ameliorated hepatic lipid deposition, ballooning degeneration, and inflammatory infiltration (Figure 1D). Moreover, steatosis score(Figure 1E), lobular inflammation score (Figure 1F), and ballooning score (Figure 1G)were much lower after folic acid intervention. The rats fed an HFD for 16 wk showed bridging fibrosis through Masson and Sirius red staining (Figure 2A). Treatment with folic acid resulted in less severe fibrosis based on the pathological sections.Furthermore, folic acid downregulated the expression levels of α-smooth muscle actin(Figure 2C), transforming growth factor beta 1 (Figure 2D), collagen type I alpha 1(Figure 2E), collagen type II alpha 1 (Figure 2F), and collagen type III alpha 1 (Figure 2G). Although folic acid could reduce the fibrosis score, the difference did not reach statistical significance (P= 0.072, Figure 2B).

Table 1 Primer sequences for real-time quantitative polymerase chain reaction
Rats in the HFD group showed significant dyslipidemia. Serum ALT (P< 0.01),AST (P< 0.01), FBG (P< 0. 01), TG (P< 0.01), TC (P< 0.01), and LDL (P< 0.01) levels were significantly elevated compared with those in the control group, accompanied by lower HDL (P< 0.01) levels (Table 2). The folic acid group showed a significant reduction in FBG (P< 0.01), TG (P< 0.01), TC (P< 0.01), and LDL (P< 0.01) levels.However, there was no significant difference in HDL levels between the HFD and folic acid groups. Abnormal bile acid metabolism and Hcy metabolism were detected in the HFD group. HFD rats had higher TBA (P< 0.01) and Hcy (P< 0.01) levels than the control group. Folic acid significantly reduced serum TBA (P< 0.05) and Hcy (P<0.01) levels compared with those in the HFD group (Table 2). The results above suggested that folic acid ameliorates HFD-induced hepatic lipid accumulation,inflammation, and fibrosis.
Folic acid inhibits hepatic lipogenesis and promotes hepatic fatty acid oxidation in rats with HFD-induced NASH
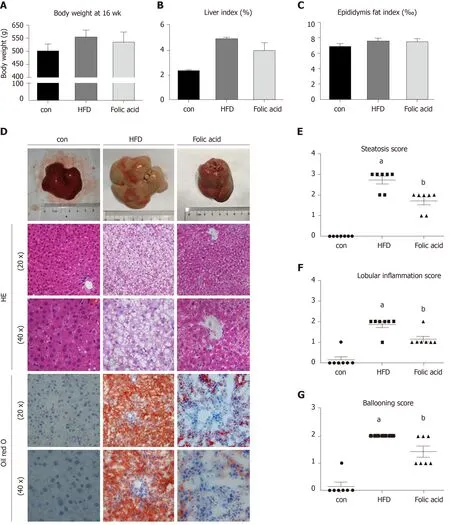
Figure 1 Folic acid ameliorates histological alterations in high-fat diet-induced steatohepatitis independent of affecting body weight. A: Body weight at 16 wk in each group; B: Liver index in each group; C: Epididymis fat index in each group; D: Hematoxylin-eosin and Oil red staining in each group. Scale bars: 50 μm; EG: Steatosis score, lobular inflammation score, and ballooning score in each group. All the data are expressed as the mean ± SE (n = 4-7). aP < 0.05 vs con group; bP< 0.05 vs HFD group. HFD: High-fat diet; HE: Hematoxylin-eosin.
Abnormal hepatic lipid uptake,de novolipogenesis (DNL), and β-oxidation contribute to the progression of NAFLD[15]. To further characterize the effects of folic acid on hepatic lipid metabolism in HFD-induced NASH rats, we analyzed the expression levels of genes related to DNL, β-oxidation, and lipid uptake. As shown in Figure 3AD, folic acid significantly downregulated the expression levels of sterol regulatory element binding transcription protein 1c, stearoyl-CoA desaturase, acetyl-CoA carboxylase, and fatty acid synthase. Moreover, genes related to hepatic lipid βoxidation and lipid uptake such as PPARγ (Figure 3E), long-chain specific acyl-CoA dehydrogenase (Figure 3F), FABP1 (Figure 3G), CPT1α (Figure 3H), and fatty acid transport protein 2 (Figure 3I) were elevated after folic acid administration. To further confirm the ameliorative effect of folic acid on hepatic lipid β-oxidation. We also detected the expression levels of related genes at the protein level. As shown in Figure 3J-L, CPT1α, and FABP1 levels were strikingly reduced by HFD and significantly restored by folic acid intervention. Furthermore, liver cholesterol (Figure 3M) and triglyceride (Figure 3N) levels were reduced in the folic acid group compared with the HFD group. This part of results suggested that folic acid improves abnormal hepatic lipid metabolism and then reduces hepatic lipid accumulation.

Figure 2 Folic acid ameliorates liver fibrosis in the rat model. A: Masson and Sirius red staining in each group. Scale bars: 100 μm; B: Fibrosis score in each group; C-G: Hepatic αSMA, TGFβ1, Col1a1, Col2a1, and Col3a1 in each group. All the data are expressed as the mean ± SE (n = 4-7). aP < 0.05 vs con group; bP <0.01 vs con group; cP < 0.01 vs HFD group. HFD: High-fat diet; αSMA: α-smooth muscle actin; TGFβ1: Transforming growth factor beta 1; Col1a1: Collagen type I alpha 1; Col2a1: Collagen type II alpha 1; Col3a1: Collagen type III alpha 1.
Folic acid restores the expression levels of PPARα via SIRT1 in rats with HFDinduced NASH and Huh7 cell line
Both PPARs and SIRT1 are key regulators in hepatic lipid β-oxidation. To further determine the effect of folic acid on the remission of hepatic β-oxidation in rats with HFD-induced NASH, we first evaluated the expression levels of SIRT1 and PPARα in animal models. As shown in Figure 4A-C, rats in the HFD group displayed lowerlevels of SIRT1 and PPARα than controls. Folic acid could strongly restore the expression levels of SIRT1 and increase the expression of PPARα to a certain extent.

Table 2 Serological lipid metabolism indexes in each group
Next, we constructed a PA-induced steatosis cell model using the Huh7 cell line. 5-MTHF, a predominant form of folic acid, was used as an intervention drug. After 12 h of treatment with PA solution, the expression levels of SIRT1 (P< 0.05) and PPARα (P< 0.05) were significantly downregulated. 5-MTHF strongly elevated the expression levels of SIRT1 (1.45-fold in the 1 μg/mL and 1.26-fold in 10 μg/mL 5-MTHF group compared with the levels in the PA treatment group, Figure 4D and F) and PPARα(1.29-fold in the 1 μg/mL and 1.44-fold in 10 μg/mL 5-MTHF group compared with the levels in the PA treatment group, Figure 4D and G). The upregulating effect of PPARα by 5-MTHF was dramatically blocked after knockdown of SIRT1 with a siRNA (Figure 4E, H, and I). Overall, folic acid restores hepatic PPARα levelsviaa SIRT1-dependent mechanism and then improves hepatic lipid metabolism under HFD-feeding conditions.
在传统的TSP问题中,解构建图为所有城市节点的连接网,边的权值为两城市节点间的距离。在本文中,首先需确定所有待编配车组与除晚高峰外的待编配车次的成本矩阵Cij,其中行代表车组号,列代表列车车次;车次按时间从早到晚依次排列,成本矩阵中的每一个值都作为一个节点vij(第i个车组担任第j个车次)。在进行计划编制时需按照车次时间顺序依次进行编配,故将所有节点按从左到右的方向依次连接,当前节点仅可与右侧相邻列的所有节点连接,如图1所示。
Folic acid improves hepatic one-carbon metabolism in rats with HFD-induced NASH
We measured the serum folic acid level in each group to further characterize the effect of the folic acid intervention. As shown in Figure 5A, folic acid intragastric administration significantly increased serum folic acid levels, although there was no difference in serum folic acid levels between the control and HFD groups. To further evaluate the effect of folic acid on one-carbon metabolism under HFD conditions, we detected the expression levels of key enzymes involved in one-carbon metabolism.qRT-PCR showed decreasedALDH1L1(0.15-fold,P< 0.01, Figure 4C) andMAT1A(0.10-fold,P< 0.01, Figure 4D) levels in the HFD group than controls. Western blot confirmed the lower MAT1A level as well (Figure 4B and 4B). Folic acid supplementation could increase the expression of those genes at the transcription(Figure 4C and D) or translation levels (Figure 4B and E). The results above implied that folic acid could partially restore depleted one-carbon metabolism in HFD rats and suggested that folic acid has a direct effect on the liver.
Folic acid restores the diversity of the gut microbiota and the gut barrier and improves endotoxemia and liver inflammation in the NASH rat model
Fecal samples were collected and subjected to 16S rRNA sequencing to detect the effect of folic acid on the gut microbiota. As shown in Figure 6A, folic acid restored the alpha diversity based on PD_whole_tree measurement, which demonstrated that folic acid could restore the HFD-induced depletion of the gut microbiota abundance.Principal coordinates analysis showed that folic acid could alter the composition of the gut microbiota in HFD-fed rats (Figure 6B). The unweighted pair-group method with arithmetic mean analysis showed that folic acid partially restored the alteration in the overall structure of the gut microbiota induced by the HFD (Figure 6C and D).Besides, compared to the control group, lower abundance of Bacteroidetes was detected in the HFD group, and folic acid administration could partially increase levels of Bacteroidetes. Compared with the HFD group, an increase in several genera such as Pseudomonadaceae and Leptotrichiaceae was observed (data not shown).Moreover, HE and Occludin immunohistochemical staining of the ileum showed that folic acid could restore the villus structure and the abundance of the expression of tight junctions (Figure 6E). Moreover, serum endotoxin levels were significantly reduced in the folic acid group (Figure 6F). Then, the expression levels of proinflammatory factors such as tumor necrosis factor alpha (Figure 6G), interleukin-6 (Figure 6H), and interleukin-1 beta (Figure 6I); chemokine receptor C-C chemokine receptor type 2 (Figure 6J); and oxidative stress-related factors such as neutrophil cytosol factor 1 (Figure 6K), neutrophil cytosolic factor 2 (Figure 6L), cytochrome b-245 alpha chain (Figure 6M), and cytochrome b-245 beta chain (Figure 6N) were greatly decreased by folic acid treatment. Overall, folic acid could restore the depleted diversity and the intestinal barrier, ameliorate endotoxemia, and decrease hepatic inflammatory reactions under HFD conditions.

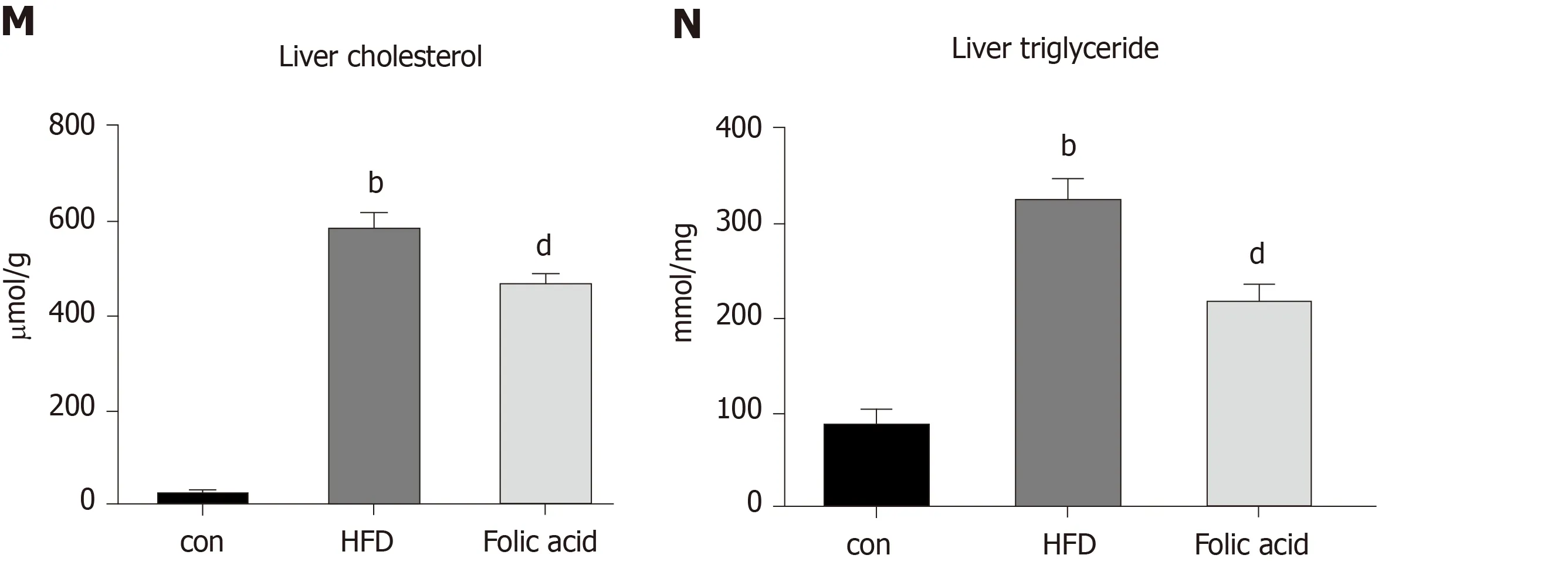
Figure 3 Folic acid inhibits hepatic lipogenesis and promotes hepatic fatty acid oxidation in high-fat diet-induced steatohepatitis rats. A-I: mRNA expression levels of SREBP1c, SCD, ACACA, FASN, PPARγ, ACADL, FABP1, CPT1α, and FATP2 in each group; J-L: Protein expression levels of CPT1α and FABP1 in each group; M and N: Liver cholesterol and triglyceride levels. All the data are expressed as the mean ± SE (n = 3-6). aP < 0.05 vs con group; bP < 0.01 vs con group; cP < 0.05 vs HFD group; dP < 0.01 vs HFD group. HFD: High-fat diet; SREBP1c: Sterol regulatory element binding transcription protein 1c; SCD: Stearoyl-CoA desaturase; ACACA: Acetyl-CoA carboxylase; FASN: Fatty acid synthase; PPARγ: Peroxisome proliferator-activated receptor gamma; ACADL: Long-chain specific acyl-CoA dehydrogenase; FABP1: Fatty acid binding protein 1; CPT1α: Carnitine palmitoyltransferase 1A; FATP2: Fatty acid transport protein 2.
DISCUSSION
We demonstrated that folic acid attenuated hepatic lipid metabolism in rats with HFD-induced steatohepatitis, increased PPARα levels through a SIRT1-dependent mechanismin vivoandvitro, ameliorated HFD-induced depleted hepatic one-carbon metabolism, and restored the diversity of the gut microbiota, thus contributing to the improvements of HFD-induced NASH in rats.
One of important findings in our present study is that folic acid plays an important role in regulating hepatic lipid metabolism in the HFD-induced NASH model. Lipid metabolism disorder is one of the most important pathophysiological changes in individuals with NAFLD. Either the “two-hit” or “multiple parallel hits” hypothesis confirms that abnormal lipid metabolism is one of the core causes of steatosis[16,17].Both increased DNL[18]and impaired fatty acid oxidation[19]contribute to the pathogenesis of NAFLD. Previous studies confirmed that folic acid could reduce lipid accumulation in primary chicken hepatocytes[12]and alter lipid metabolism genes in male rat offspring[20]. Studies also indicated that folic acid may alleviate abnormal lipid metabolism and cholesterol deposition in the liver through the LKB1-AMPK pathway[11]. However, the further mechanism for the effect of folic acid in regulating hepatic fatty acid oxidation is still rarely known. PPARs belong to the nuclear hormone receptor superfamily, and of the PPARs, PPARα regulates hepatic lipid metabolism, glucose metabolism, and liver inflammation[21,22]. Numerous rate-limited enzymes associated with fatty acid uptake[23]and mitochondrial β-oxidation[24,25]are regulated by PPARα. Hepatocyte-specific PPARα deletion impaired fatty acid homeostasis and promoted the progression of NAFLD[26]. SIRT1 is an NAD+-dependent deacetylase in mammalian cells that plays a key role in metabolic diseases[27]and regulates the transcription network in free fatty acid oxidation[28].Microarray analysis confirmed that SIRT1, PPARα, and peroxisome proliferatoractivated receptorγcoactivator-1 (PGC1α) played a core role in the regulation of genes responsible for β-oxidation[29]. Hepatic deletion of SIRT1 could impair PPARα signaling, and overexpression of SIRT1 could restore the expression levels of PPARα and its target genes[30]. We confirmed that folic acid restores hepatic PPARαviaa SIRT1 dependent pathway, which further reveals the effect of folic acid on hepatic lipid metabolism.

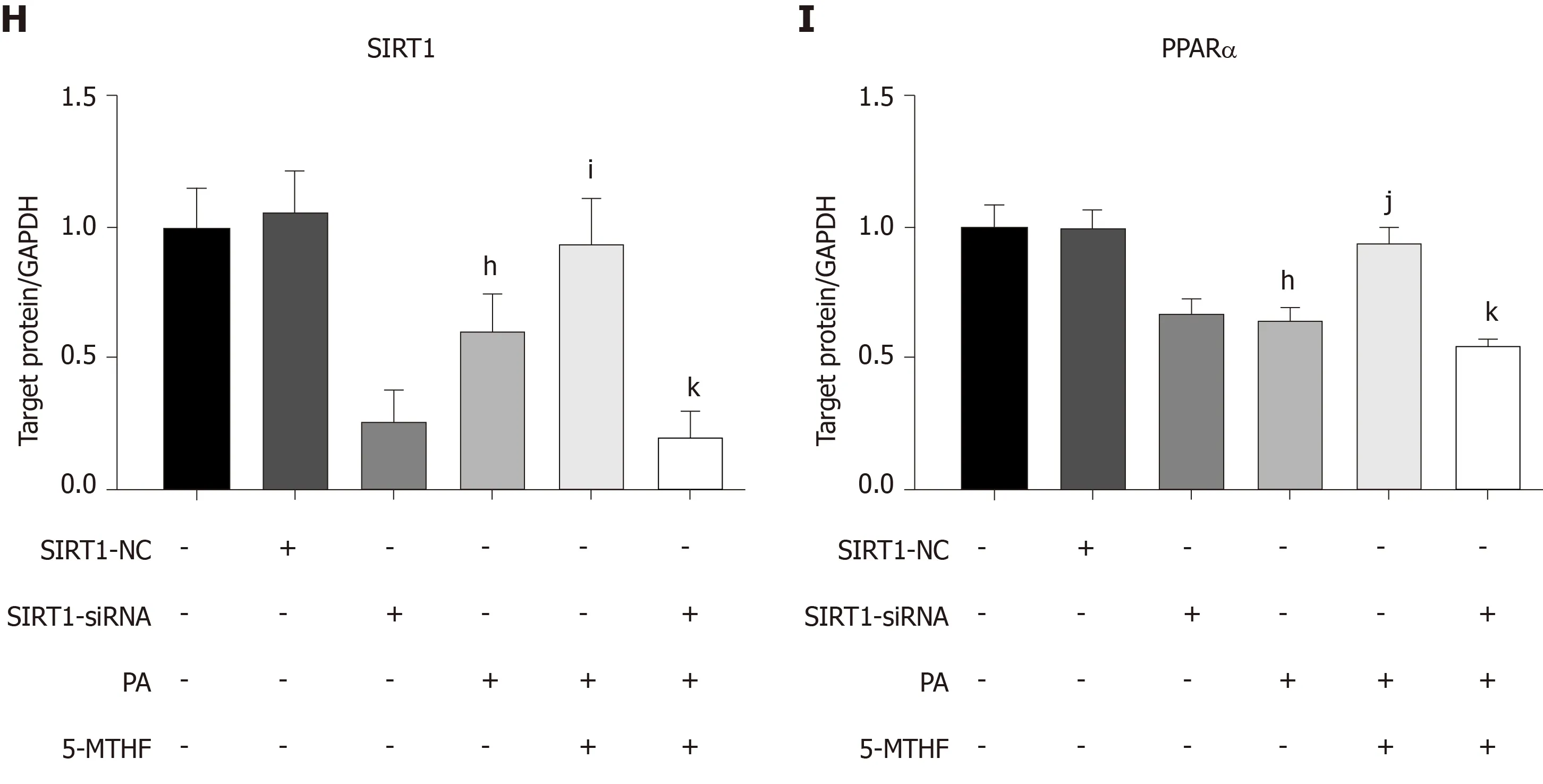
Figure 4 Folic acid restores the expression levels of PPARα via SlRT1 in rats with high-fat diet-induced steatohepatitis and Huh7 cell line. A-C: The expression levels of SIRT1 and PPARα in each group of rats; D, F, and G: The expression levels of SIRT1 and PPARα in Huh7 cell line exposed to 0.3 mmol/L PA; E,H, and I: The expression levels of SIRT1 and PPARα in Huh7 cell line transfected with SIRT1 siRNA and then exposed to 0.3 mmol/L PA. All the data are expressed as the mean ± SE (n = 3). aP < 0.05 vs con group; bP< 0.01 vs con group; dP < 0.01 vs HFD group; eP < 0.05 vs control; fP < 0.05 vs 0.3 PA group; gP < 0.01 vs 0.3 PA group; hP < 0.01 vs SIRT1-NC group; iP < 0.05 vs 0.3 PA group; jP < 0.01 vs 0.3PA group; kP <0.01 vs 5-MTHF and 0.3 PA group. HFD: High-fat diet; PPARα:Peroxisome proliferator-activated receptor alpha; SIRT1: Silence information regulation factor 1; PA: Palmitic acid; 5-MTHF: 5-methyltetrahydrofolic acid.
Significantly lower FBG levels in the folic acid group indicated that folic acid may play a role in glucose metabolism in metabolic diseases including NAFLD. Studies showed that chronic folic acid deficiency induced glucose metabolism disorder[31].Folic acid treatment decreased serum glucose levels in a diabetic rat model[32].Administration of folic acid improved insulin resistance by altering the DNA methylation profile in HFD-fed mice[33]. This indicated that folic acid could improve glucose metabolism in NASH conditions, but specific mechanisms need further research.
Another finding in our present study is that folic acid could restore one-carbon metabolism in rats with HFD-induced NASH. Several studies have reported that folic acid and other methyl donors have an alleviating effect on chronic liver diseases, such as liver fibrosis[34], cholestasis[34], drug-induced liver injury[6,35], alcoholic liver disease[13], obesity[36], and NAFLD[37,38]. However, serum folic acid level in NAFLD patients is still controversial in recent studies. Some researchers[39]believed that there was no significant difference in serum folic acid and vitamin B12 levels between NAFLD and healthy control groups and that neither folic acid nor vitamin B12 levels were associated with pathological severity. Other studies have found varying degrees of positive results. Hirschet al[40]found a lower serum folic acid concentration in female obese patients with NAFLD than in healthy controls; Mahamidet al[41]posited that lower folate or vitamin B12 levels were associated with the histological severity of NASH. An association between serum folic acid levels and the severity of liver steatosis was also found by research on Chinese adult NAFLD patients[42]. The serum folic acid levels in the NAFLD patients from the abovementioned literature varied from 9.3 to 12.6 ng/mL on average, all of which were normal levels. Therefore, we believed that HFD had little effect on folate absorption or serum folate levels. This result was consistent with the lack of a significant difference in serum folic acid levels between the control and HFD groups in our study. However, as a co-enzymatic substrate, folic acid serves a core role in on-carbon transfer reactions. Folic aciddependent one-carbon metabolism is important for methylation reactions in mammal cells[43]. It has been well demonstrated that differential DNA methylation occurs in individuals with NAFLD[44-46]. Genes involved in one-carbon metabolism showed abnormal DNA methylation, and of these genes, MAT1A and ALDH1L1 showed hypermethylated levels and downregulation[46]. MAT1A participates in the synthesis of S-adenosylmethionine[47]and ALDH1L1 is involved in metabolism in the carbon pathway[48]. Both of them are required for lipid homeostasis. We found strong downregulation of MAT1A and ALDH1L1 in HFD-fed rats in our present study, and additional folic acid supplementation was effective in restoring their expression levels. These findings indicated that folic acid supplementation is required for NASH individuals to improve hepatic lipid metabolism through restoring one-carbon metabolism.

Figure 5 Folic acid improves hepatic one-carbon metabolism in rats with high-fat diet-induced steatohepatitis. A: Serum folic acid levels in each group; C and D: mRNA levels of MAT1A and ALDH1L1 in each group; B and E: Protein levels of MAT1A in each group. All the data are expressed as the mean ± SE (n = 3-8). aP <0.05 vs con group bP < 0.01 vs con group; cP < 0.05 vs HFD group; dP < 0.01 vs HFD group. HFD: High-fat diet; MAT1A: Methionine adenosyltransferase 1A.
An increasing number of studies have demonstrated that NAFLD has a diseasespecific gut microbiome signature[49], of which the depleted diversity of the microbiota and microbial gene richness and differential bacterial clusters were most commonly reported[50,51]. Additionally, HFD consumption disturbed gut permeability by reducing tight-junction proteins such as Occludin and ZO-1, which leads to endotoxemia and chronic systemic inflammation[52]and promotes the progression of NASH. We notably found that folic acid could stabilize the intestinal barrier and the diversity of the gut microbiota, which partially explained the calming effect on the whole body and hepatic inflammation.
There are still some limitations that deserve further study. First, we have not demonstrated the effect of folic acid on the SIRT1-PPARα pathwayin vivo. SIRT1 conditional knock-out mice should be used in future study to further evaluate the molecular mechanism of folic acid in the improvement of NAFLD. Second, studies have confirmed that several genes related to lipid metabolism, such asPGC1α[53],ZNF274, andSREBP2[44], had enriched DNA methylation in individuals with NAFLD.Therefore, whether folic acid could influence the balance of acetylation and methylation in genes related to free fatty acid oxidation, especiallyPPARαandPGC1α,remains an interesting question. Finally, a drug-dose gradientin vivocould be considered to evaluate the optimal intervention dose for clinical guidance.
In conclusion, we have confirmed the improvement effect of folic acid on HFDinduced NASH in rats. We demonstrated that folic acid improves hepatic lipid metabolism by upregulating PPARαviaa SIRT1-dependent mechanism. Meanwhile,folic acid administration restores depleted hepatic one-carbon metabolism and the diversity of gut microbiota in HFD-fed rats. These results further clarify the therapeutic role of folic acid in NAFLD and its possible mechanism, suggesting that folic acid may become a therapeutic drug to treat NAFLD in the future.


Figure 6 Folic acid restores the diversity of the gut microbiota and the gut barrier and improves endotoxemia and liver inflammation in a non-alcoholic steatohepatitis rat model. A: Alpha diversity of the gut microbiota; B: Principal coordinates analysis; C and D: Unweighted pair-group method with arithmetic mean analysis; E: Hematoxylin-eosin and Occludin immunochemical staining of the ileum. Scale bars: 100 μm; F: Serum endotoxin levels in each group; G-N: Hepatic TNFα, Il-6, IL-2β, CCR2, p47phox, p67phox, p22phox, and gp91phox levels in each group. All the data are expressed as the mean ± SE (n = 4-6). aP < 0.05 vs con groupbP < 0.01 vs con group; cP < 0.05 vs HFD group; dP < 0.01 vs HFD group. HFD: High-fat diet; HE: Hematoxylin-eosin; TNF-α: Tumor necrosis factor alpha; IL:Interleukin; CCR2: Chemokine receptor C-C chemokine receptor type 2.
ARTICLE HIGHLIGHTS
Research background
Non-alcoholic fatty liver disease has become a global burden, but there is still a lack of convinced drug therapy strategies for non-alcoholic steatohepatitis (NASH). As one of essential watersoluble vitamins for the human body, folic acid may become one of the drug targets for treatment of NASH, but the specific mechanism is not fully understood.
Research motivation
As one of essential vitamins absorbed by the intestine mainly, food-sourced folic acid improved high-fat diet (HFD)-induced steatohepatitis in previous studies, but further mechanism of folic acid on host hepatic lipid metabolism and the effect of folic acid on lipid one-carbon metabolism and gut microbiota remains rarely understood.
Research objectives
We aimed to evaluated the effect of folic acid on HFD-fed rat models and further clarify the mechanism of folic acid on hepatic lipid metabolism and gut microbiota.
Research methods
An HFD-induced rat model of NASH was used in the present study. Treatment of folic acid by oral administration lasted for 8 wk. Hepatic lipid metabolism was evaluated by real-time quantitative polymerase chain reaction (qRT-PCR). Expression levels of silence information regulation factor 1 (SIRT1) and peroxisome proliferator-activated receptor alpha (PPARα) were measured by Western blot analysis in HFD-induced rat models and palmitic acid-induced Huh7 cells. SIRT1 siRNA was transfected in Huh7 cells to examine whether folic acid restored PPARα levels through a SIRT1-dependent mechanism. Genes and proteins related to hepatic one-carbon metabolism were detected by qRT-PCR and Western blot. 16S rDNA sequencing was used to evaluate the effect of folic acid on gut microbiota profile.
Research results
Administration of folic acid ameliorated HFD-induced steatohepatitis. Folic acid repaired impaired hepatic lipid β-oxidation and hepatic one-carbon metabolism. SIRT1 and PPARα levels were restored by folic acid treatment. The restoration effect of PPARα by folic acid was blocked after SIRT1 knockdownin vitro. Furthermore, folic acid restored the diversity and altered the overall structure of gut microbiota profile.
Research conclusions
Folic acid restores PPARα levelsviaa SIRT1-dependent mechanism, ameliorates HFD-induced impaired hepatic lipid metabolism and hepatic one-carbon metabolism, and improves the diversity of gut microbiota, thus acting a protective role in HFD-induced NASH in rats.
Research perspectives
Folic acid may become one of drug targets for treatment of NASH. Research about folic acid in epigenetic regulation may further clarify the mechanism of folic acid on NASH.
猜你喜欢
杂志排行

World Journal of Gastroenterology的其它文章
- Medical management of metabolic and cardiovascular complications after liver transplantation
- New role for ceramide in hypoxia and insulin resistance
- Role of gut microbiota on intestinal barrier function in acute pancreatitis
- Prognostic significance of hepatic encephalopathy in patients with cirrhosis treated with current standards of care
- Optimal proximal resection margin distance for gastrectomy in advanced gastric cancer
- Computed tomography vs liver stiffness measurement and magnetic resonance imaging in evaluating esophageal varices in cirrhotic patients: A systematic review and meta-analysis