阿拉善马鹿冬季肠道菌群的多样性
2020-02-21李俊乐滕丽微刘振生
李俊乐 高 惠 滕丽微,2 刘振生,2*
(1.东北林业大学野生动物与自然保护地学院,哈尔滨,150040;2.国家林业和草原局野生动物保护学重点开放实验室,哈尔滨,150040)
肠道菌群(gut microorganism)是动物肠道内复杂的,具有动态变化的微生物群落[1]。肠道菌群从动物一出生就开始在其肠道繁殖,其种类繁多且复杂多样,经过长期的进化,宿主的肠道中形成一个复杂且动态平衡的体系[2]。在动物肠道中,肠道菌群的数量与种类并不固定,外界环境和宿主自身都会对上述动态平衡产生影响,从而影响肠道微生物的多样性,肠道菌群与多种外界因素相互作用共同影响宿主的消化吸收、免疫调节及生理健康等方面[3-4]。肠道菌群是宿主快速适应外界环境的重要因素。研究表明食草动物独特的肠道为植物纤维素降解形成一个独特的环境,而草食动物的肠道菌群具有特殊的代谢特点,能分泌大量的纤维素分解酶类[5-6]。反刍动物更是如此,这种独特的肠道菌群和肠道有助于宿主适应高纤维食物[7]。因此反刍动物肠道菌群与其他大多数动物肠道菌群相比,有更突出的作用[8]。随着近年来学者开始对动物肠道微生物的菌群结构,菌群多样性等方面进行深入研究[9-10]。野生哺乳动物肠道菌群的研究已经从简单的菌群组成分析深入到其功能分析,但对肠道微生物群落中的功能菌及其机制仍有待解决。因此,对肠道菌群的组成和多样性的研究仍是重要内容。
马鹿(Cervuselaphus)是国家Ⅱ级重点保护野生动物[11],中国主要有阿拉善亚种(C.alashanicus)、塔里木亚种(C.e.yarkandensis)、天山亚种(C.e.songari-cus)、阿尔泰亚种(C.e.sibiricus)、东北亚种(C.e.xanthopygus)、甘肃亚种(C.e.kansuensis)、四川亚种(C.e.macneilli)和西藏亚种(C.e.wallichii)8个亚种[12]。目前阿拉善马鹿仅分布于宁夏和内蒙古交界的贺兰山中段,是我国唯一幸存的该亚种的有效种群。刘振生等[13-14]和骆颖等[15]已对贺兰山地区阿拉善马鹿的生境选择进行了较为详细的研究,乔付杰等[16]对该地区野生阿拉善马鹿分子学进行研究。但目前关于野生阿拉善马鹿肠道微生物的研究尚未见报道。因此,本研究通过对阿拉善马鹿肠道菌群进行研究,探索其个体间肠道菌群多样性和菌群结构之间的差异,对阿拉善马鹿更科学合理的保护提供理论支持。
1 研究地概况
贺兰山位于宁夏回族自治区与内蒙古自治区交界处(38°21′—39°22′N,105°49′—106°42′E),分属于内蒙古自治区贺兰山国家级自然保护区和宁夏贺兰山国家级自然保护区。处于北温带向荒漠过渡地带,是一条重要的自然地理分界线[17]。贺兰山地处内陆,是典型的大陆性季风气候,夏季较短,冬季漫长。贺兰山日照时间较长,年平均日照时数为2 800—3 000 h。贺兰山海拔较高,平均海拔在2 000—3 000 m。贺兰山地区各月份降水量有明显差异,最低降水量和最高降水量分别为187.6 mm和627.5 mm,年平均降水量为420 mm[18-19]。
2 材料与方法
2.1 样品采集
2017年2—4月初在贺兰山地区采集野生阿拉善马鹿群体的粪便样本98份。采集粪便样品时为防止交叉感染,每采集一个样本,换一副一次性PE手套。将采集的粪便样本放入95%的无水乙醇中保存,运回实验室后放入-80℃冰箱保存。
2.2 个体鉴定
本实验使用QIAGEN QIAamp Fast DNA Stool Mini Kit试剂盒提取DNA,实验方法详见说明书。将提好的DNA分成两份,一份放置于-20 ℃,用于下一步扩增,一份放置于-80 ℃冻存备用。用10对高度多态的微卫星引物(RT1、BM888、HAUT14、CSSM19、T156、BMS1248、T530、DM45、CSRM602、IOBT9651)对所有样本DNA进行扩增,进行个体识别,抽样时排出3对重复个体。最终选取20份DNA质量较高的粪便样品,进行后续分析。
样品编号分别为WA1、WA2、WA3、WA4、WA5、WN1、WN2、WN3、WN4、WN5、WA6、WA7、WA8、WA9、WA10、WN6、WN7、WN8、WN9、WN10。
2.3 高通量测序
利用高通量测序技术,在保证样品浓度一致和测序量相同的条件下,在Miseq 2×300 bp平台上,对20份阿拉善马鹿样品进行16S rRNA的V3-V4区进行扩增。将得到的序列去除嵌合体和靶向区域外序列,得到有效序列。然后,将具有高度相似性(97%)的有效序列进行OUT划分,并绘制韦恩图。通过与数据库的对比,进行物种分类分析,得到样品所具有的菌群。Alpha 多样性通过Ace、Chao1、Shannon和Simpsoon 4个指标来衡量菌群得丰富性,绘制稀释曲线来验证实验样品数量的合理性。一般来说,ACE和Chao1指数越大,说明群落的丰富度越高,Shannon指数越大菌群的丰富度越高,Simpsoon指数越大,菌群的丰富度越低。主成分分析(PCA)法,是以样本间差异性最大的两个因素分别为横纵坐标绘制PCoA聚类图。样品间距离越近,则说明样本间菌群相似性越高。
3 结果与分析
3.1 样品中的序列和OTU数目
本研究通过高通量测序技术对20份样品便进行16S RNA测序。共获得1 564 112条序列,有效序列1 399 562条,平均每个样品中含有78 205.6条序列,69 978.1条有效序列。基于相似度>97%的原则,将获得的有效tags序列进行聚类,共获得2 740个OTUs,平均每个样品含有137个OTUs(表1)。其中,有效序列数量最高的是WA4有107 484条有效序列,最低的是WN7有37 569条有效序列,二者相差69 915条。韦恩图中重叠部分为所有样品共有的OUT数目,由图2可知20个样品中共有110个OTUs。
3.2 Alpha 多样性分析
通过Alpha 多样性分析指数对20份样品进行丰富度和多样性比较。从图2可以看出,随着测试深度的不断加深,稀释曲线从不断上升,直至趋于平缓,说明样品的测序深度比较合理。
Alpha多样性分析指数分析结果说明(表2),样品菌群的丰富度和多样性都相对较高。20份样品中菌群丰富度指数相对较高的为WN2(Ace 3 132.50,Chao1 2 750.16)和WA1(Ace 3 028.42,Chao1 2 580.29),相对较低的为WA3(Ace 2 106.89,Chao1 2 011.40)和WA5(Ace 2 217.13,Chao1 1 892.97)。样品中Shannon指数相对较高的为WN7(5.82)和WN8(5.85),相对较低的WA5(4.27)。样品中Simpsoon指数相对较高的为WA5(0.06)和WN1(0.04),相对较低的为WN8(0.009)。样品的覆盖度都在98.48%以上,说明测序结果足以充分地反映了样品的真情况。
表1 样品中有效序列及OUT数量
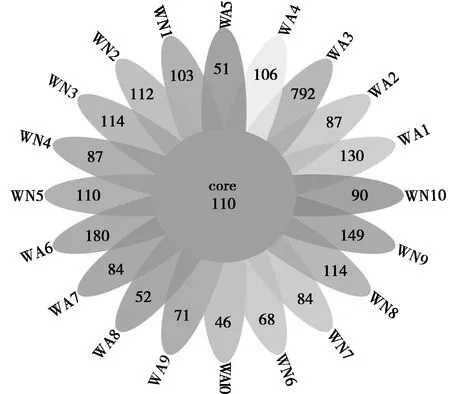
图1 OTUs韦恩图Fig.1 Venn diagram of OTUs
表2 马鹿肠道微生物多样性指数
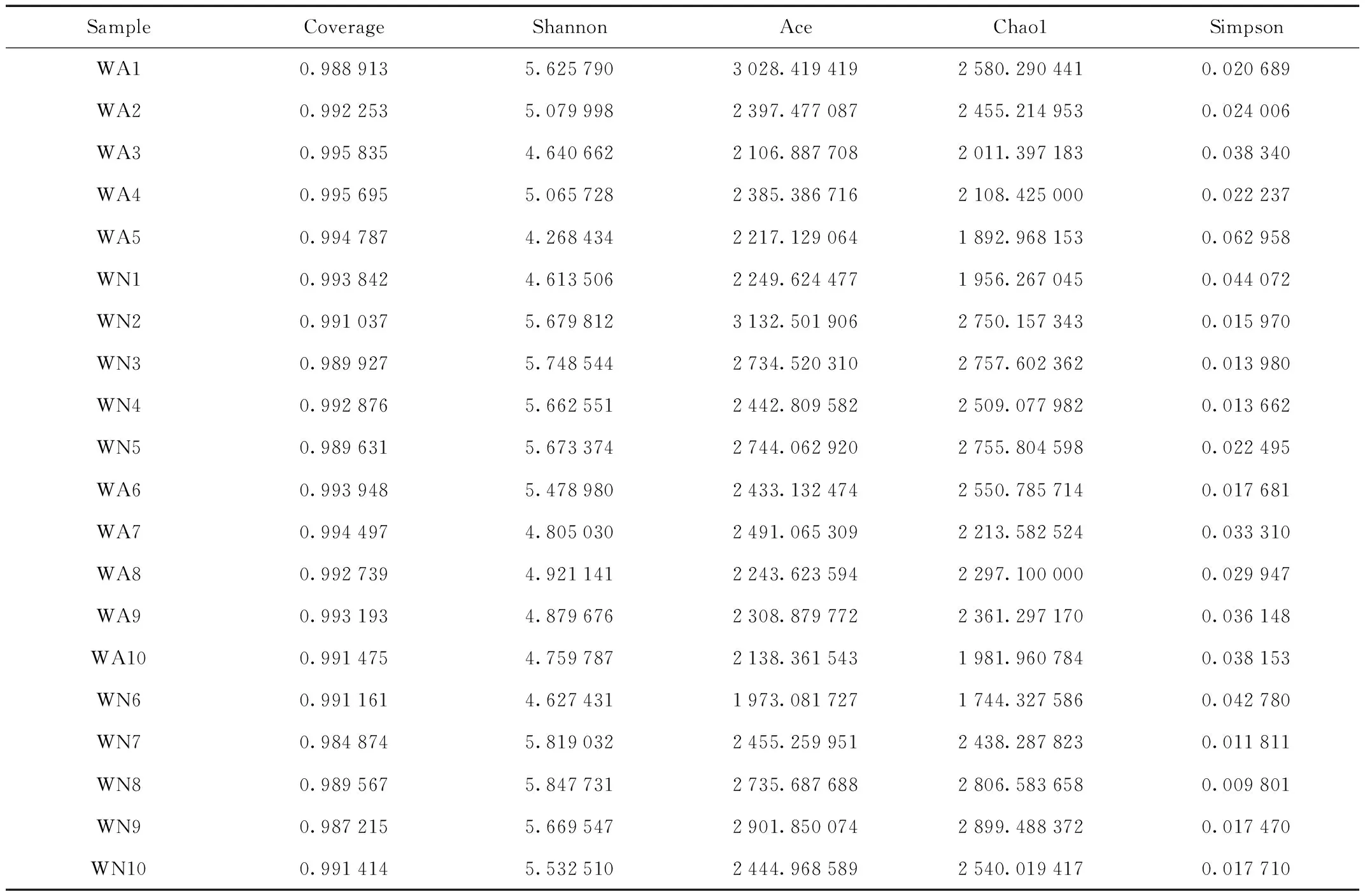
Tab.2 Intestinal microbial diversity index of red deer
3.3 物种注释
在门水平和属水平上对样品进行物种注释,20个样品共获得24个菌门,283个菌属。
在门水平上,含量大于1%的菌门有5个(图3),分别是厚壁菌门(Firmicutes),(76.71±14.71)%、拟杆菌门(Bacteroidetes),(14.16±7.92)%、变形菌门(Proteobacteria),(3.51±10.38)%、疣微菌门(Verrucomicrobia),(1.92±2.37)%、放线菌门(Actinobacteria),(1.33±2.14)%。
在属水平上,含量大于1%的菌属有11个(图4),分别为孢子杆菌属(Sporobacter),(7.38±3.61)%、拟杆菌属(Bacteroides),(4.15±1.67)%、梭菌属系列(ClostridiumIV、ClostridiumXlV),(3.63±1.44)%、罗氏菌属(Roseburia),(3.00±1.74)%、胃瘤球菌(Ruminococcus),(1.85±0.93)%、奥氏杆菌(Oscillibacter),(1.80±0.93)%、Intestinimonas(1.56±0.88)%、Akkermansia(1.50±2.88)%、Alistipes(1.49±1.11)%、Flavonifractor(1.47±1.02)%。
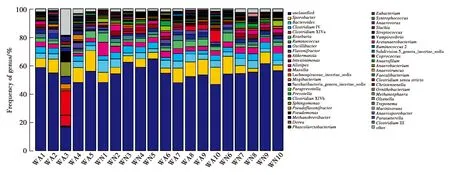
图4 阿拉善马鹿属水平菌群相对丰度Fig.4 Relative microbial abundance composition at the genus level
3.4 主成分分析
根据样本OUT的种类及其相对丰度,对样本进行主成分分析,绘制PCoA聚类图。样本间越聚集,说明样本间相似度越高,微生物菌群结构相似度越高。由图5可知,除样品WA10和WA3外,其余样品聚集程度很高,说明样品间菌群差别不大。
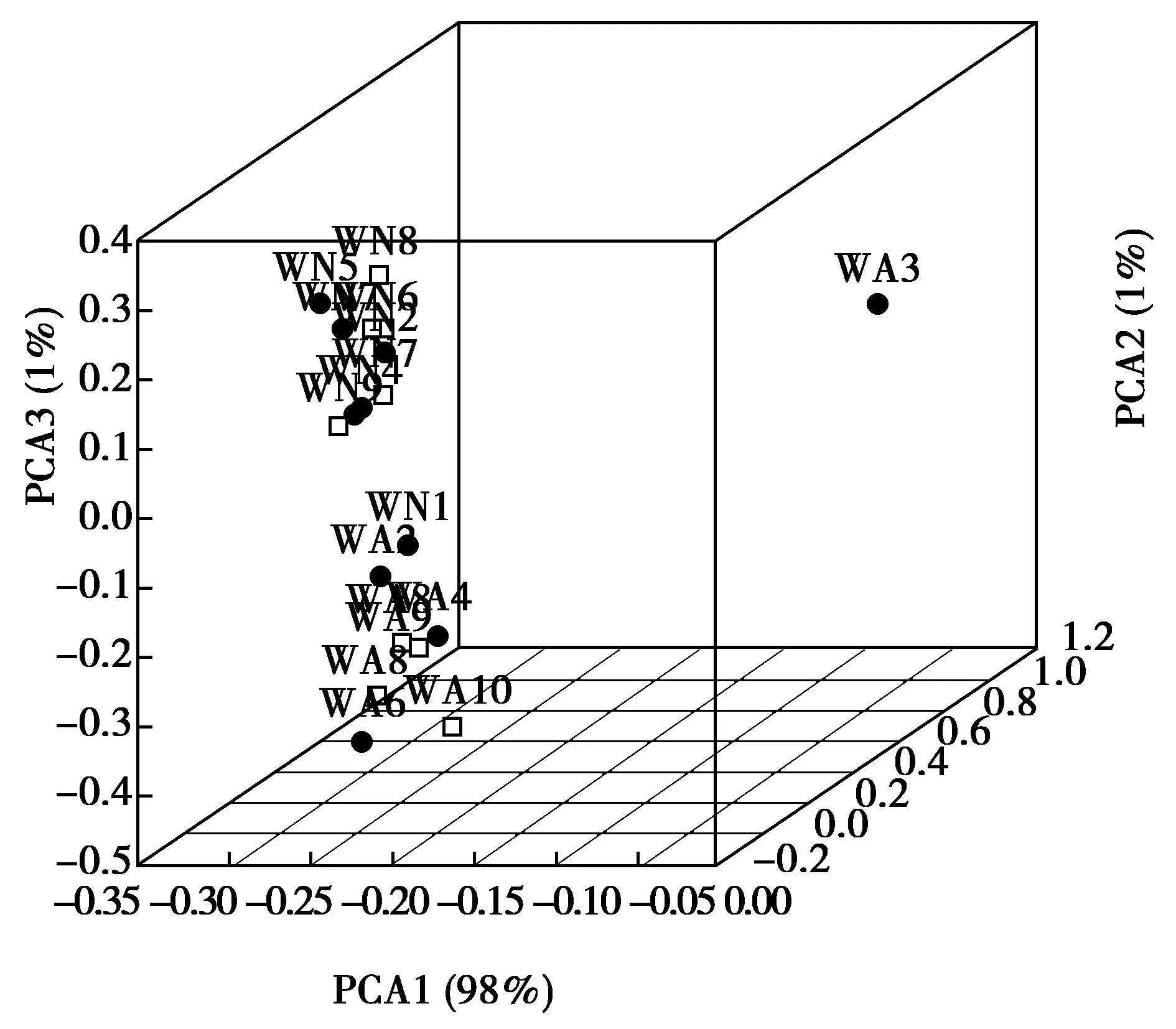
图5 PCA主成分分析Fig.5 Principal component analysis of PCA
4 讨论
与传统技术相比高通量测序技术能较全面地反应样品中微生物的种类。因此,本文采用16S rRNA高通量测序技术来分析阿拉善马鹿肠道菌群的构成。已有研究表明成年川金丝猴(Rhinopithecusroxellana)肠道菌群的核心菌门为厚壁菌门和拟杆菌门[20];大熊猫(Ailuropodamelanoleuca)肠道菌群的优势菌门为厚壁菌门、变形菌门和梭杆菌门(Fusobacteria)[21];美洲獾(Taxideataxus)肠道菌群的核心菌门为厚壁菌门、拟杆菌门、放线菌门和疣微菌门[22];海南坡鹿(Cervuseldiihainanus)肠道菌群的核心菌门为厚壁菌门、拟杆菌门和变形菌门[23],这些研究表明,不同的物种其肠道菌群存在一定的差异。
实验数据表明,20只阿拉善马鹿的优势肠道菌群与其他动物的优势肠道菌群基本相同,主要为厚壁菌门和拟杆菌门。厚壁菌门是马鹿肠道菌群中含量最高的菌群。厚壁菌门能够降解食物中的纤维并将分解产物提供给宿主使用,同时食用高纤维食物也有利于厚壁菌门的积累[24]。拟杆菌门主要功能是帮助宿主降解碳水化合物、蛋白质等物质,维持肠道菌群平衡,还可增强宿主免疫性[25-26]。研究发现,肥胖者体内的厚壁菌门比瘦者多,而拟杆菌比瘦者少。随后的一年时间里,将肥胖者的食物改为低脂肪低碳水化合物后,发现他们的体重均有降低,同时厚壁菌的含量降低,拟杆菌的含量升高,说明厚壁菌可以帮助机体从食物吸取更多的能量[27]。而作为反刍动物的野外马鹿,需要对纤维、淀粉等物质进行吸收利用,为自身提供必要的能量。但反刍动物的肠胃本身并不能产生相应的酶类,不能对其进行降解。因此,肠道菌群对降解食物中的纤维和淀粉起到了重要的作用。
肠道菌群多样性与性别、年龄和食性都有一定的关系[28-29],因此性别、年龄和食性都会是造成其差异的原因。对阿拉善马鹿肠道菌群多样性比较发现,除WA10和WA3外,阿拉善马鹿肠道菌群多样性无明显差别,但其肠道菌群含量占比有一定差别。可能是由于马鹿个体间体积差异,对食物的需求量也不同,从而形成了肠道菌群多样性的差异。研究发现,同域野外马麝(Moschuschrysogaster)的核心菌群主要为厚壁菌门和拟杆菌门[30]。且二者在食性上有很大程度的重叠[31]。就野外环境来说,冬季天气寒冷,食物匮乏,阿拉善马鹿和马麝需要消耗大量的能量来逃避捕猎者,维持体温等。而肠道菌群会帮助它们从食物中汲取能量维持身体所需。由此可见,食性会对动物的代谢等方面产生影响,并能改变动物的肠道菌群[29]。本实验所用到的粪便样品数量较少,对阿拉善马鹿肠道菌群的更详细规律有待进一步研究。
